
Identification of a Branch Number Locus in Soybean Using BSA-Seq and GWAS Approaches
 , ,
, ,
Abstract
:1. Introduction
2. Results
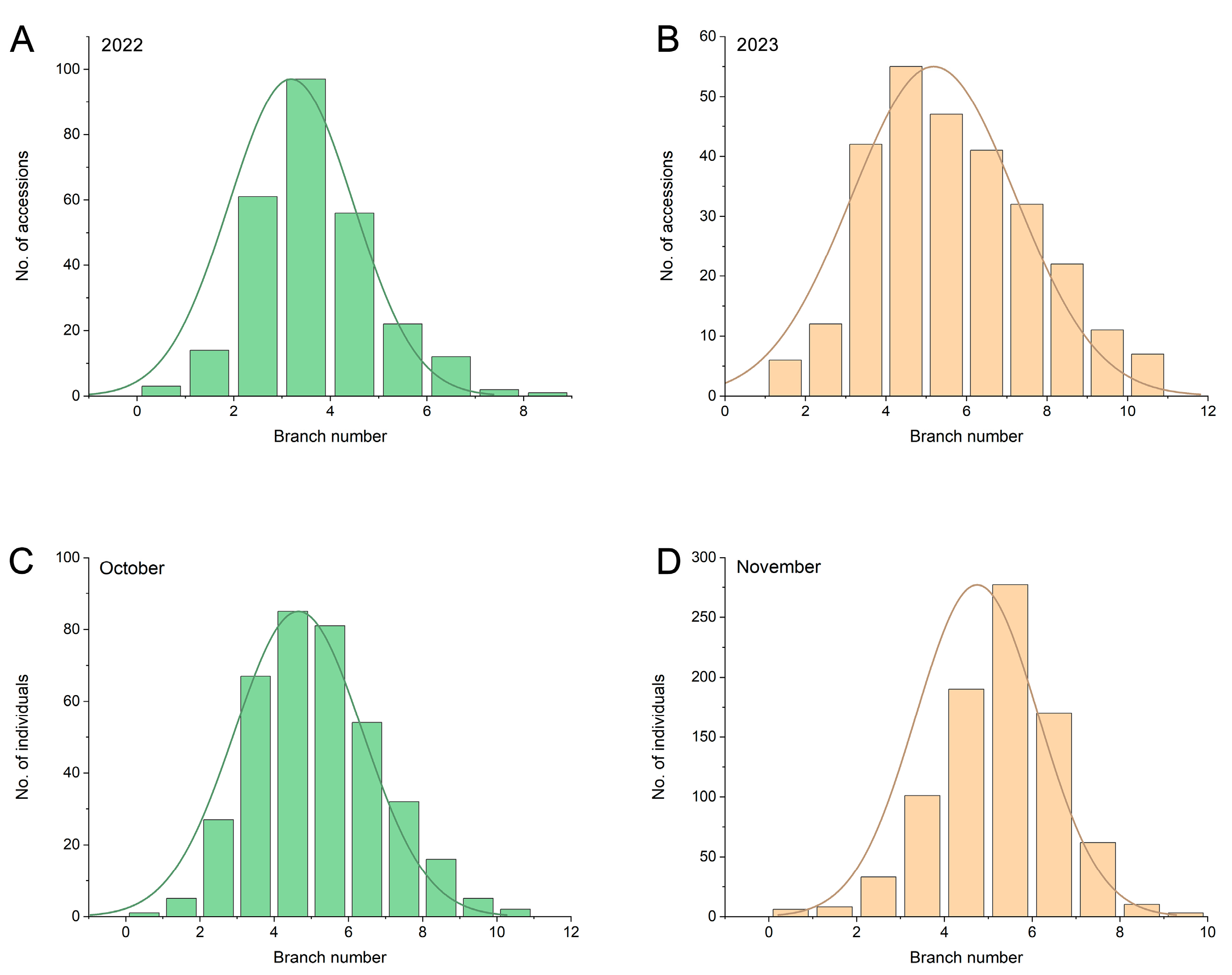
2.1. Evaluation of Branch Number in the Association Panel and F2 Population
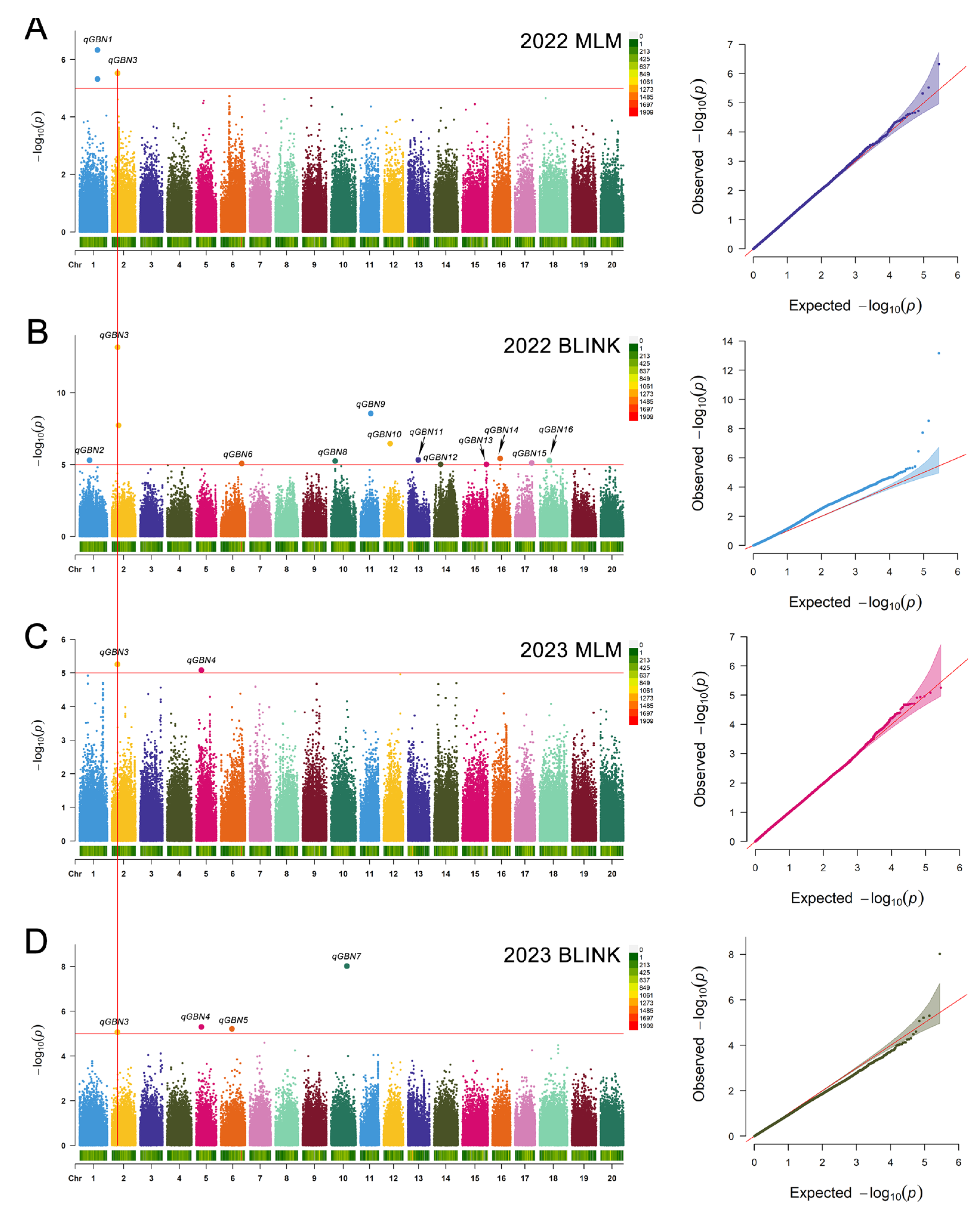
2.2. Genomic Regions of Branch Number Identified by GWAS
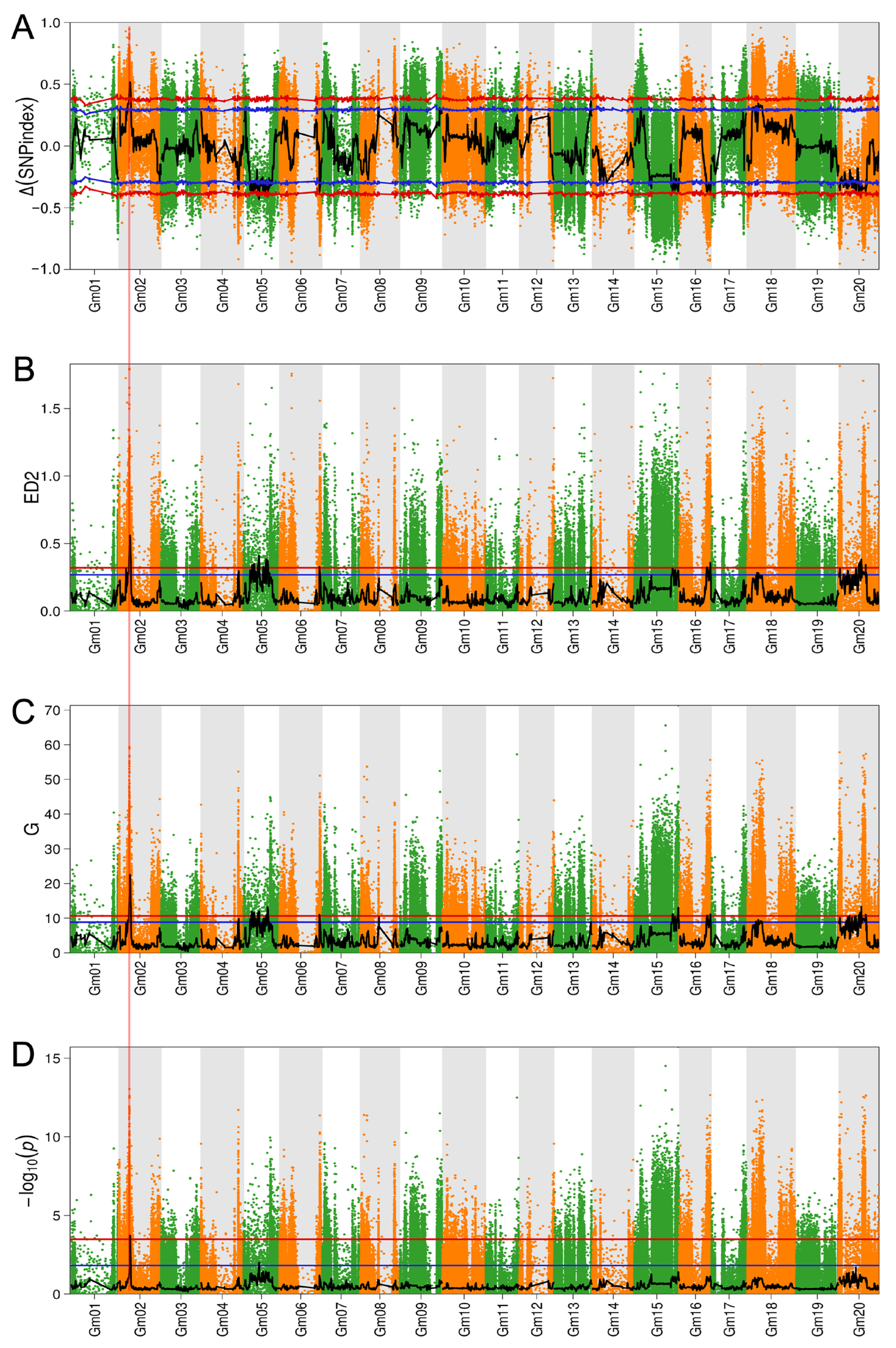
2.3. BSA-Seq-Based Identification of Branch Number-Associated Genomic Regions
2.4. Candidate Loci Identified by GWAS and BSA
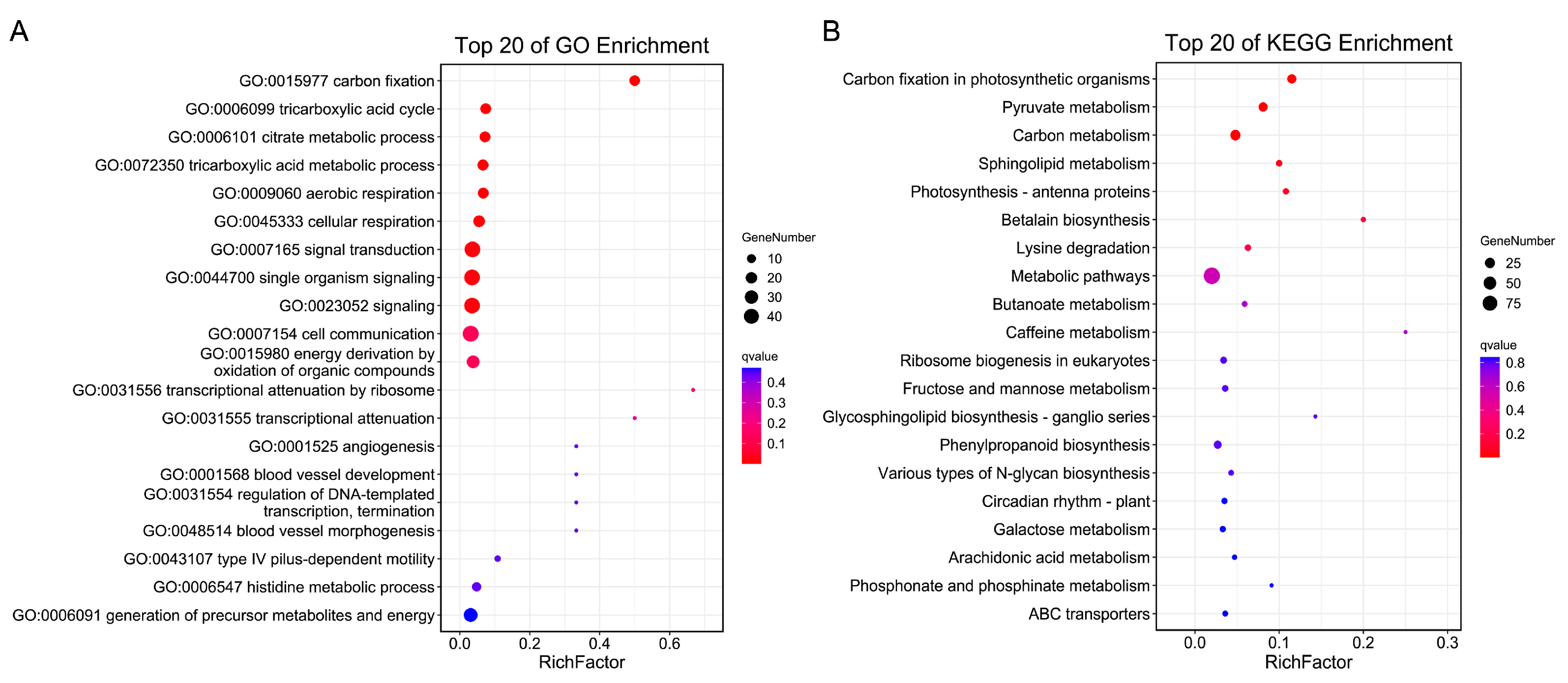
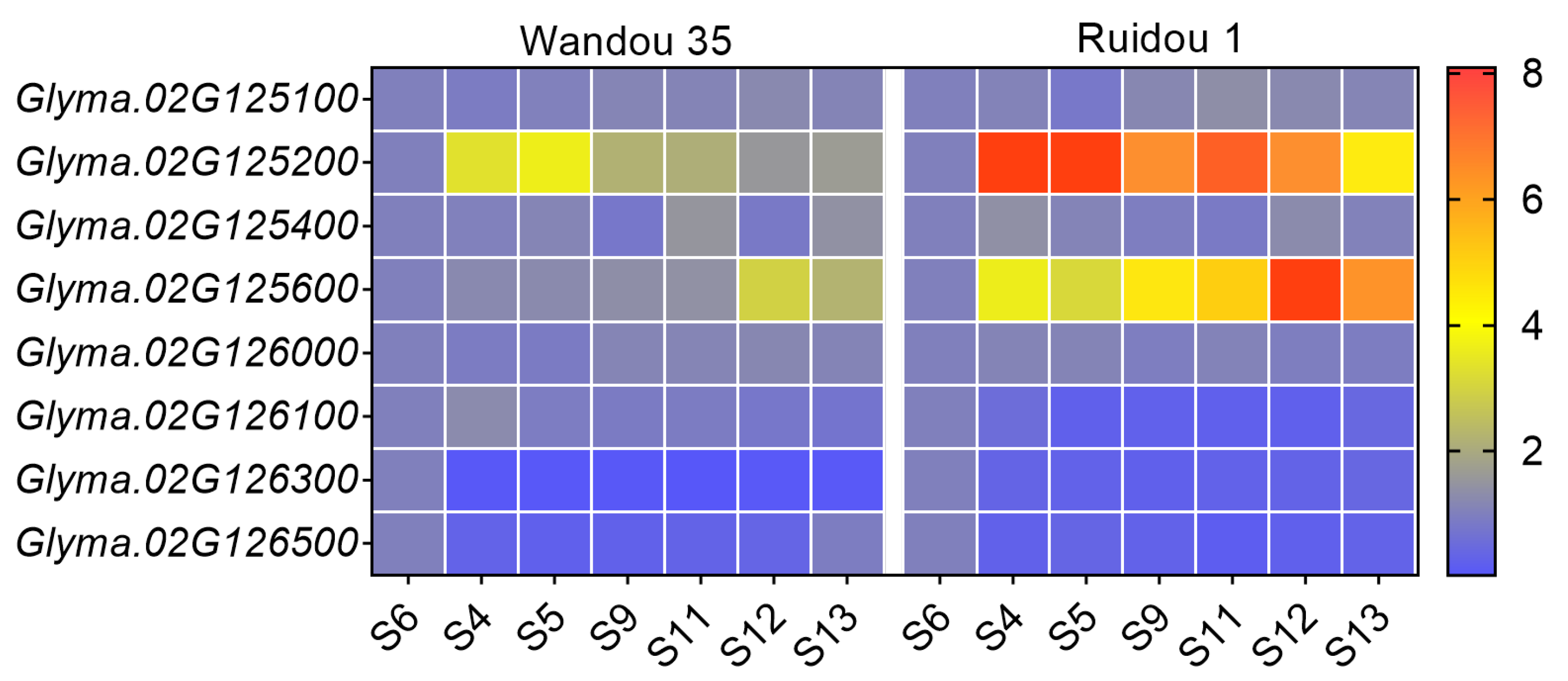
2.5. Candidate Gene Annotation and Expression Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Phenotypic Evaluation
4.2. Statistical Analysis
4.3. DNA Extraction and Whole-Genome Resequencing
4.4. Genome-Wide Association Analyses
4.5. Bulk Segregant Sequencing Analyses
4.6. Candidate Genes Identification and Description
4.7. Analyses of Gene Expression Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean—Worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield Trends Are Insufficient to Double Global Crop Production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Caballero-Anthony, M.; Lassa, J.; Nair, T. Towards Asia 2025: Policy and technology imperatives. Summary of the main findings of the second international conference on Asian food security held in Singapore on 21–22 August 2014. Food Secur. 2015, 7, 159–165. [Google Scholar] [CrossRef]
- Zakaria, N.H.; Nordin, M.S.; Ibrahim, M.A.; Abdul Majid, F.A.; Zainuddin, Z. Genetic diversity and proteomic analysis of vegetable soybean (Glycine max (L.) Merrill) accessions grown in mineral and BRIS soils. Czech J. Genet. Plant Breed. 2023, 59, 14–22. [Google Scholar] [CrossRef]
- Pedersen, P.; Lauer, J.G. Soybean growth and development in various management systems and planting dates. Crop Sci. 2004, 44, 508–515. [Google Scholar] [CrossRef]
- Shim, S.; Kim, M.Y.; Ha, J.; Lee, Y.-H.; Lee, S.-H. Identification of QTLs for branching in soybean (Glycine max (L.) Merrill). Euphytica 2017, 213, 225. [Google Scholar] [CrossRef]
- Agudamu; Yoshihira, T.; Shiraiwa, T. Branch development responses to planting density and yield stability in soybean cultivars. Plant Prod. Sci. 2016, 19, 331–339. [Google Scholar] [CrossRef]
- Modi, M.R.; Katochi, M.; Thakur, N.; Gautam, M.; Chodhary, S.; Chahota, R.K. Mapping of genomic regions associated with dwarfing and the determinate growth habit in horsegram (Macrotyloma uniflorum). Czech J. Genet. Plant Breed. 2023, 59, 196–204. [Google Scholar] [CrossRef]
- Kulkarni, S.R.; Balachandran, S.M.; Ulaganathan, K.; Balakrishnan, D.; Praveen, M.; Prasad, A.S.H.; Fiyaz, R.A.; Senguttuvel, P.; Sinha, P.; Kale, R.R.; et al. Molecular mapping of QTLs for yield related traits in recombinant inbred line (RIL) population derived from the popular rice hybrid KRH-2 and their validation through SNP genotyping. Sci. Rep. 2020, 10, 13695. [Google Scholar] [CrossRef]
- Yin, S.; Li, P.; Xu, Y.; Xue, L.; Hao, D.; Liu, J.; Yang, T.; Yang, Z.; Xu, C. Logistic model-based genetic analysis for kernel filling in a maize RIL population. Euphytica 2018, 214, 86. [Google Scholar] [CrossRef]
- Hu, J.; Wang, X.; Zhang, G.; Jiang, P.; Chen, W.; Hao, Y.; Ma, X.; Xu, S.; Jia, J.; Kong, L.; et al. QTL mapping for yield-related traits in wheat based on four RIL populations. Theor. Appl. Genet. 2020, 133, 917–933. [Google Scholar] [CrossRef]
- Chen, Q.-S.; Zhang, Z.-C.; Liu, C.-Y.; Xin, D.-W.; Qiu, H.-M.; Shan, D.-P.; Shan, C.-Y.; Hu, G.-H. QTL Analysis of Major Agronomic Traits in Soybean. Agric. Sci. China 2007, 6, 399–405. [Google Scholar] [CrossRef]
- Sayama, T.; Hwang, T.-Y.; Yamazaki, H.; Yamaguchi, N.; Komatsu, K.; Takahashi, M.; Suzuki, C.; Miyoshi, T.; Tanaka, Y.; Xia, Z.; et al. Mapping and comparison of quantitative trait loci for soybean branching phenotype in two locations. Breed. Sci. 2010, 60, 380–389. [Google Scholar] [CrossRef]
- Yang, Y.; Lei, Y.; Bai, z.; Wei, Y.; Zhang, R. QTL Mapping and integration as well as candidate gene prediction for branch number in soybean. bioRxiv 2019. [Google Scholar] [CrossRef]
- Ran, H.; Rongxia, G.; Zhangxiong, L.; Xiaoli, Z.; Ruzhen, C.; Lijuan, Q. Mapping the qBN-c1-1 Locus to LG C1 for Soybean Branching Using Residual Heterozygous Lines Derived from a Segregation Population. Sci. Agric. Sin. 2009, 42, 1152–1157. [Google Scholar]
- Zhongwen, H.; Tuanjie, Z.; Deyue, Y.; Shouyi, C.; Junyi, G. Detection of QTLs of Yield Related Traits in Soybean. Sci. Agric. Sin. 2009, 42, 4155–4165. [Google Scholar]
- Zhu-li, Y.; Gui-quan, L.I. The QTL Analysis of Important Agronomic Traits on a RIL Population from a Cross between Jinda52 and Jinda57. Acta Agric. Boreali-Sin. 2010, 25, 88–92. [Google Scholar]
- Yao, D.; Liu, Z.Z.; Zhang, J.; Liu, S.Y.; Qu, J.; Guan, S.Y.; Pan, L.D.; Wang, D.; Liu, J.W.; Wang, P.W. Analysis of quantitative trait loci for main plant traits in soybean. Genet. Mol. Res. 2015, 14, 6101–6109. [Google Scholar] [CrossRef]
- Zhang, H.; Hao, D.; Sitoe, H.M.; Yin, Z.; Hu, Z.; Zhang, G.; Yu, D. Genetic dissection of the relationship between plant architecture and yield component traits in soybean (Glycine max) by association analysis across multiple environments. Plant Breed. 2015, 134, 564–572. [Google Scholar] [CrossRef]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef]
- Shim, S.; Ha, J.; Kim, M.Y.; Choi, M.S.; Kang, S.-T.; Jeong, S.-C.; Moon, J.-K.; Lee, S.-H. GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). Int. J. Mol. Sci. 2019, 20, 135. [Google Scholar] [CrossRef]
- Lamlom, S.F.; Zhang, Y.; Su, B.; Wu, H.; Zhang, X.; Fu, J.; Zhang, B.; Qiu, L.-J. Map-based cloning of a novel QTL qBN-1 influencing branch number in soybean [Glycine max (L.) Merr.]. Crop J. 2020, 8, 793–801. [Google Scholar] [CrossRef]
- Liang, Q.; Chen, L.; Yang, X.; Yang, H.; Liu, S.; Kou, K.; Fan, L.; Zhang, Z.; Duan, Z.; Yuan, Y.; et al. Natural variation of Dt2 determines branching in soybean. Nat. Commun. 2022, 13, 6429. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.-A.; Peng, D.-L.; et al. Global Dissection of Alternative Splicing in Paleopolyploid Soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. Sowing Date and Planting Density on Growth and Development of Spring Soybean in Southern Xinjiang. Master’s Thesis, Tarim University, Xinjiang, China, 2023. [Google Scholar]
- Wu, Y.; Wang, E.; Gong, W.; Xu, L.; Zhao, Z.; He, D.; Yang, F.; Wang, X.; Yong, T.; Liu, J.; et al. Soybean yield variations and the potential of intercropping to increase production in China. Field Crop Res. 2023, 291, 108771. [Google Scholar] [CrossRef]
- Ghodrati, G. Study of genetic variation and broad sense heritability for some qualitative and quantitative traits in soybean (Glycine max L.) genotypes. Agric. Food Sci. 2013, 2, 31–35. [Google Scholar]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Komatsu, M.; Maekawa, M.; Shimamoto, K.; Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 Genes Determine the Inflorescence Architecture of Rice by Controlling Rachis-Branch and Spikelet Development. Dev. Biol. 2001, 231, 364–373. [Google Scholar] [CrossRef]
- Staswick, P.E.; Serban, B.; Rowe, M.; Tiryaki, I.; Maldonado, M.n.T.; Maldonado, M.C.; Suza, W. Characterization of an Arabidopsis Enzyme Family That Conjugates Amino Acids to Indole-3-Acetic Acid. Plant Cell 2005, 17, 616–627. [Google Scholar] [CrossRef]
- Zou, X.; Long, J.; Zhao, K.; Peng, A.; Chen, M.; Long, Q.; He, Y.; Chen, S. Overexpressing GH3.1 and GH3.1L reduces susceptibility to Xanthomonas citri subsp. citri by repressing auxin signaling in citrus (Citrus sinensis Osbeck). PLoS ONE 2019, 14, e0220017. [Google Scholar] [CrossRef]
- Weiste, C.; Pedrotti, L.; Selvanayagam, J.; Muralidhara, P.; Fröschel, C.; Novák, O.; Ljung, K.; Hanson, J.; Dröge-Laser, W. The Arabidopsis bZIP11 transcription factor links low-energy signalling to auxin-mediated control of primary root growth. PLOS Genet. 2017, 13, e1006607. [Google Scholar] [CrossRef] [PubMed]
- E, Z.G.; Zhang, Y.P.; Zhou, J.H.; Wang, L. Roles of the bZIP gene family in rice. Genet. Mol. Res. 2014, 13, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience 2018, 8, giy154. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLOS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Ren, W.; Zhao, L.; Liang, J.; Wang, L.; Chen, L.; Li, P.; Liu, Z.; Li, X.; Zhang, Z.; Li, J.; et al. Genome-wide dissection of changes in maize root system architecture during modern breeding. Nat. Plants 2022, 8, 1408–1422. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinf. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic Mapping and Mutant Identification without Crossing in Rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.-C.; Yost, H.J. MMAPPR: Mutation Mapping Analysis Pipeline for Pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Liu, X.; Wang, R.; Zhai, C.; Peng, G.; Yu, F.; Fernando, W.G.D. Fine mapping of Brassica napus blackleg resistance gene Rlm1 through bulked segregant RNA sequencing. Sci. Rep. 2019, 9, 14600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Panthee, D.R. Next-generation sequencing-based bulked segregant analysis without sequencing the parental genomes. G3—Genes Genom. Genet. 2021, 12, jkab400. [Google Scholar] [CrossRef]
- He, L.; Li, M.; Chen, D.D.; Qian, Q.; Zeng, D.L.; Zhu, L. Rice Ferredoxins localize to chloroplasts/plastids and may function in different tissues. Plant Signal. Behav. 2021, 16, 1926813. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Mean | SD | CV (%) | Min | Max | Kurt | Skew |
|---|---|---|---|---|---|---|---|
| 2022 | 3.18 | 1.28 | 40.25 | 0.17 | 8.33 | 0.93 | 0.64 |
| 2023 | 5.20 | 2.07 | 39.82 | 0.67 | 10.17 | 0.64 | 0.93 |
| QTL | SNP | Chromosome | Position | p Value | R2 | Allelic Variation | Year | Method |
|---|---|---|---|---|---|---|---|---|
| qGBN1 | S01_647060 | 1 | 37500028 | 4.69 × 10−7 | 11.65 | C/T | 2022 | MLM |
| S01_647058 | 1 | 37499994 | 4.79 × 10−6 | 9.82 | T/C | 2022 | MLM | |
| qGBN2 | S11_383602 | 1 | 20996571 | 5.00 × 10−6 | 9.51 | T/A | 2022 | BLINK |
| qGBN3 | S02_1240704 | 2 | 12264757 | 6.94 × 10−14 | 27.18 | A/G | 2022 | BLINK |
| S02_1241353 | 2 | 12324064 | 1.87 × 10−8 | 14.17 | C/T | 2022 | BLINK | |
| S02_1240704 | 2 | 12264757 | 2.99 × 10−6 | 9.77 | 2022 | MLM | ||
| S02_1240704 | 2 | 12264757 | 5.56 × 10−6 | 9.32 | 2023 | MLM | ||
| S02_1240704 | 2 | 12264757 | 8.57 × 10−6 | 9.25 | 2023 | BLINK | ||
| qGBN4 | S05_3893779 | 5 | 11122603 | 5.02 × 10−6 | 9.55 | T/C | 2023 | BLINK |
| S05_3893779 | 5 | 11122603 | 8.31 × 10−6 | 9.27 | 2023 | MLM | ||
| qGBN5 | S06_4772142 | 6 | 23576358 | 6.07 × 10−6 | 9.66 | C/A | 2023 | BLINK |
| qGBN6 | S06_5137235 | 6 | 44380411 | 8.54 × 10−6 | 9.25 | G/A | 2022 | BLINK |
| qGBN7 | S10_8203320 | 10 | 33337627 | 9.46 × 10−9 | 14.86 | G/T | 2023 | BLINK |
| qGBN8 | S10_7804544 | 10 | 8551686 | 5.73 × 10−6 | 9.30 | A/T | 2022 | BLINK |
| qGBN9 | S11_8849037 | 11 | 22593147 | 2.81 × 10−9 | 15.85 | T/A | 2022 | BLINK |
| qGBN10 | S12_9388376 | 12 | 13737593 | 3.51 × 10−7 | 12.17 | C/T | 2022 | BLINK |
| qGBN11 | S13_10274380 | 13 | 21682594 | 4.83 × 10−6 | 9.82 | G/A | 2022 | BLINK |
| qGBN12 | S14_10921879 | 14 | 13946264 | 9.64 × 10−6 | 9.25 | G/A | 2022 | BLINK |
| qGBN13 | S15_12533199 | 15 | 50770118 | 9.67 × 10−6 | 9.16 | A/G | 2022 | BLINK |
| qGBN14 | S16_12903158 | 16 | 16529439 | 3.82 × 10−6 | 10.02 | G/C | 2022 | BLINK |
| qGBN15 | S17_13967003 | 17 | 35344266 | 7.99 × 10−6 | 9.27 | C/A | 2022 | BLINK |
| qGBN16 | S18_14535964 | 18 | 20984058 | 5.30 × 10−6 | 9.51 | A/G | 2022 | BLINK |
| QTLs | Chromosome | Start Position (bp) | End Position (bp) | Peak | Method |
|---|---|---|---|---|---|
| qBBN1 | Gm02 | 11420001 | 15020000 | 0.559684 | ED |
| Gm02 | 11440001 | 15070000 | 22.53829 | Gst | |
| Gm02 | 11530001 | 15020000 | 0.517047 | Δ(SNP-index) | |
| Gm02 | 12280001 | 13360000 | 0.003283 | Fisher | |
| qBBN2 | Gm05 | 8580001 | 10660000 | 11.91817 | Gst |
| Gm05 | 9270001 | 10430000 | 0.340816 | ED | |
| qBBN3 | Gm05 | 11450001 | 12970000 | 11.65613 | Gst |
| Gm05 | 11460001 | 12850000 | 0.348614 | ED | |
| Gm05 | 11570001 | 12570000 | −0.37708 | Δ(SNP-index) | |
| qBBN4 | Gm05 | 16210001 | 18710000 | 11.9553 | Gst |
| Gm05 | 16210001 | 18940000 | 0.411016 | ED | |
| Gm05 | 16210001 | 18910000 | −0.4334 | Δ(SNP-index) | |
| qBBN5 | Gm05 | 22600001 | 23850000 | −0.37968 | Δ(SNP-index) |
| Gm05 | 22830001 | 23880000 | 0.329834 | ED | |
| qBBN6 | Gm05 | 25240001 | 27230000 | 0.383032 | ED |
| Gm05 | 25740001 | 26750000 | −0.39135 | Δ(SNP-index) | |
| Gm05 | 26070001 | 27230000 | 10.96294 | Gst | |
| qBBN7 | Gm05 | 27540001 | 29280000 | 0.374127 | ED |
| Gm05 | 27850001 | 29330000 | 13.24474 | Gst | |
| Gm05 | 27850001 | 29220000 | −0.41289 | Δ(SNP-index) | |
| qBBN8 | Gm06 | 47020001 | 48270000 | 11.02317 | Gst |
| Gm06 | 47060001 | 48240000 | 0.326973 | ED | |
| qBBN9 | Gm13 | 43170001 | 44200000 | 0.325159 | ED |
| qBBN10 | Gm15 | 44910001 | 46480000 | 11.18481 | Gst |
| qBBN11 | Gm15 | 51360001 | 53140000 | 13.02159 | Gst |
| qBBN12 | Gm16 | 32020001 | 33890000 | 0.324992 | ED |
| Gm16 | 32490001 | 34130000 | 10.81712 | Gst | |
| qBBN13 | Gm16 | 36050001 | 37470000 | 0.35877 | ED |
| Gm16 | 36170001 | 37440000 | 11.15172 | Gst | |
| qBBN14 | Gm20 | 19840001 | 20840000 | −0.38348 | Δ(SNP-index) |
| qBBN15 | Gm20 | 23160001 | 25850000 | 0.361823 | ED |
| Gm20 | 23330001 | 24470000 | 10.97986 | Gst | |
| qBBN16 | Gm20 | 24620001 | 25850000 | 11.71612 | Gst |
| Gm20 | 24620001 | 25660000 | −0.39685 | Δ(SNP-index) | |
| qBBN17 | Gm20 | 25860001 | 27730000 | 0.381719 | ED |
| Gm20 | 26020001 | 27610000 | 13.35137 | Gst | |
| Gm20 | 26170001 | 27320000 | −0.39762 | Δ(SNP-index) | |
| qBBN18 | Gm20 | 31420001 | 32420000 | 11.04967 | Gst |
| Gm20 | 31420001 | 32420000 | 0.341994 | ED |
| GeneID | Start Position (bp) | End Position (bp) | Symbol | Annotation |
|---|---|---|---|---|
| Glyma.02G125100 | 12220107 | 12224403 | SRG1 | 2-oxoglutarate/Fe(II)-dependent dioxygenase-like |
| Glyma.02G125200 | 12226179 | 12230868 | BHLH49 | Transcription factor bHLH49 isoform X1 |
| Glyma.02G125300 | 12229902 | 12230859 | - | - |
| Glyma.02G125400 | 12246600 | 12253822 | WIT2 | WPP domain-interacting tail-anchored protein 2-like isoform X4 |
| Glyma.02G125500 | 12268030 | 12268302 | - | - |
| Glyma.02G125551 | 12279258 | 12279410 | - | - |
| Glyma.02G125600 | 12302237 | 12304986 | GH3.1 | Indole-3-acetic acid-amido synthetase GH3.1 |
| Glyma.02G125700 | 12306697 | 12309236 | - | - |
| Glyma.02G125800 | 12314438 | 12315067 | - | - |
| Glyma.02G125900 | 12317269 | 12318465 | - | - |
| Glyma.02G126000 | 12321542 | 12323547 | IRT2 | Fe(2+) transport protein 1 |
| Glyma.02G126100 | 12368440 | 12371047 | BZIP43 | Basic leucine zipper transcription factor-like protein |
| Glyma.02G126200 | 12375750 | 12376682 | - | - |
| Glyma.02G126300 | 12388195 | 12394795 | CPN60B4 | RuBisco large subunit-binding protein subunit beta |
| Glyma.02G126500 | 12412320 | 12413154 | SNAT2 | Serotonin N-acetyltransferase 2, chloroplastic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, D.; Huang, L.; Zhang, X.; Zhang, S.; Yuan, Y.; Wu, G.; Hou, Y.; Yuan, X.; Chen, X.; Xue, C. Identification of a Branch Number Locus in Soybean Using BSA-Seq and GWAS Approaches. Int. J. Mol. Sci. 2024, 25, 873. https://doi.org/10.3390/ijms25020873
Dai D, Huang L, Zhang X, Zhang S, Yuan Y, Wu G, Hou Y, Yuan X, Chen X, Xue C. Identification of a Branch Number Locus in Soybean Using BSA-Seq and GWAS Approaches. International Journal of Molecular Sciences. 2024; 25(2):873. https://doi.org/10.3390/ijms25020873
Chicago/Turabian StyleDai, Dongqing, Lu Huang, Xiaoyan Zhang, Shiqi Zhang, Yuting Yuan, Gufeng Wu, Yichen Hou, Xingxing Yuan, Xin Chen, and Chenchen Xue. 2024. "Identification of a Branch Number Locus in Soybean Using BSA-Seq and GWAS Approaches" International Journal of Molecular Sciences 25, no. 2: 873. https://doi.org/10.3390/ijms25020873