

Epigenetic Dysregulation in the Pathogenesis of Systemic Lupus Erythematosus

Abstract
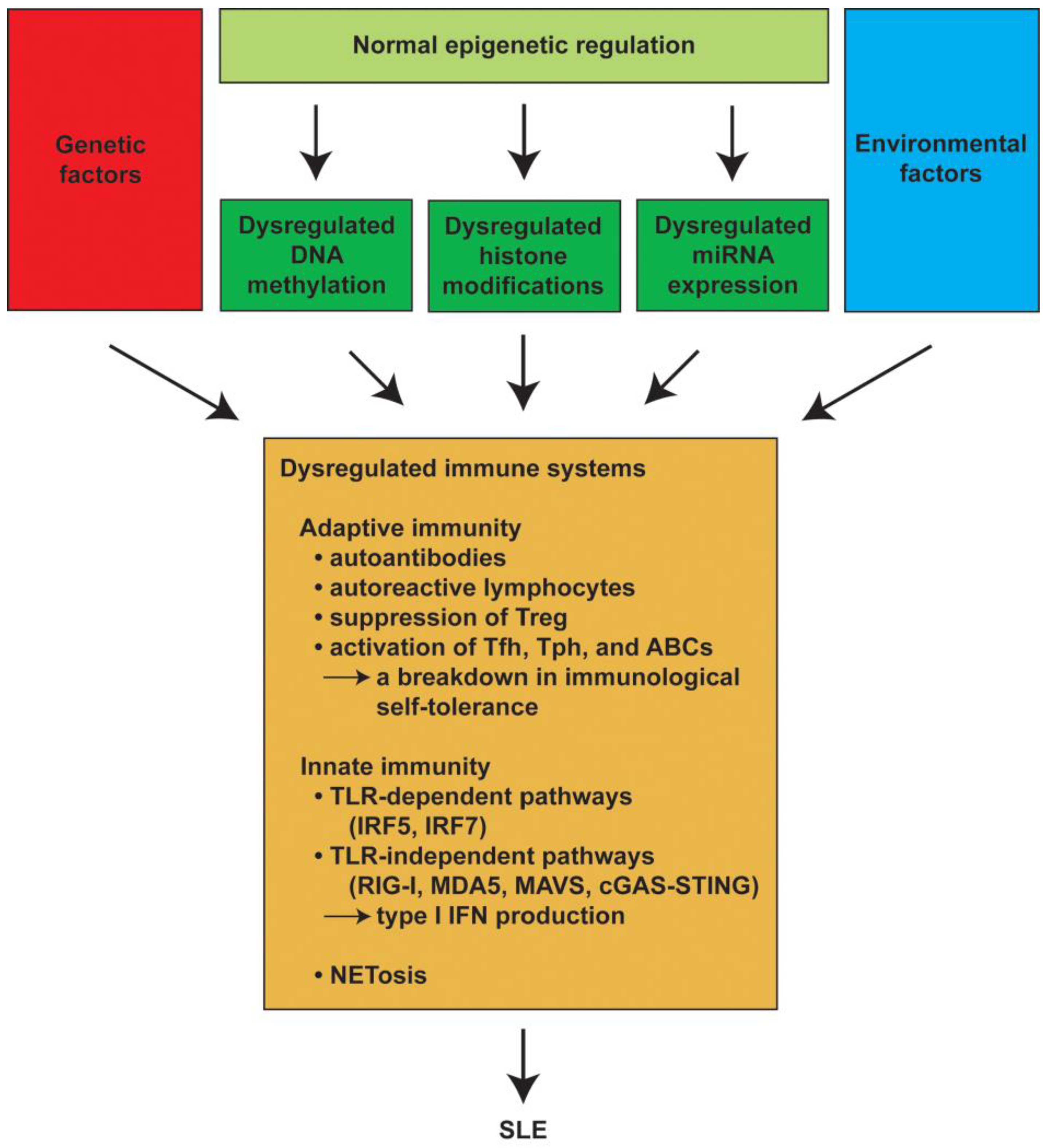
:1. Introduction
2. The Pathogenesis of SLE
2.1. Adaptive Immune Responses in SLE
2.2. Innate Immune Responses in SLE
3. Epigenetic Regulation of Chromatin Structure and Gene Transcription
4. Epigenetic Dysregulation in SLE
4.1. Dysregulated DNA Methylation in SLE
4.2. Dysregulated Histone Modifications in SLE
4.3. Dysregulated miRNA Expression in SLE

{kind=link}
| Genes | DNA Methylation | Effects | Types of Experiments | Cells | Refs. |
|---|---|---|---|---|---|
| CD11a/CD18 | hypomethylation | leukocyte adhesion and migration in inflamed tissues | in vitro | T cells | [73] |
| CD11a/CD18 | hypomethylation | leukocyte adhesion and migration in inflamed tissues | in vitro | CD4+ T cells | [78,81,87] |
| CD70 | hypomethylation | B cell activation | in vitro | CD4+ T cells | [75,78,81,87,91] |
| CD40LG | hypomethylation | B cell maturation | in vitro | CD4+ T cells | [77] |
| IL-10 | hypomethylation | B cell activation | in vitro | T cells | [79] |
| MX1, BST2, STAT1, TRIM22, IFIT1, IFIT3, IFI44L, USP18 | hypomethylation | type I IFN-mediated responses | in vitro | naïve CD4+ T cells | [83] |
| S1PR3, CD300LB, NLRP2 | hypomethylation | in vitro | CD4+ T cells | [84] | |
| IL-17A | hypomethylation | recruitment of monocytes and neutrophils | in vitro | T cells | [89] |
| PARP9, IFITM1, IFI44L | hypomethylation | type I IFN-mediated responses | in vitro | CD4+ T cells, B cells, granulocytes, monocytes | [93] |
| MX1, IFI44L, PARP9, DT3XL, IFIT1, IFI44, RSAD2, PLSCR1, IRF7 | hypomethylation | type I IFN-mediated responses | in vitro | PBMCs | [95] |
| IFI44, IFITM1, YBX1, TAF8 | hypomethylation | in vitro | B cells | [96] | |
| SOX12, ARFGAP3, CCDC81, MEG3 | hypermethylation | in vitro | B cells | [96] | |
| BCL6 | hypomethylation | Tfh cell differentiation | in vitro | naïve CD4+ T cells | [97] |
| IL-2 | hypermethylation | Treg suppression | in vitro | T cells | [106] |
| Genes | Histone Modifications | Effects | Types of Experiments | Cells | Refs. |
|---|---|---|---|---|---|
| WDR5, SLC24A3 | high H3K4me3 | in vitro | PBMCs | [98] | |
| PTPN22, METTL16, LRP1B, CDH13 | low H3K4me3 | in vitro | PBMCs | [98] | |
| CD70 | high H3K4me2, high H3ac | B cell activation | in vitro | CD4+ T cells | [99] |
| TNFα | high H4ac | inflammatory responses | in vitro | monocytes | [100] |
| IL-17 | high H3ac | recruitment of monocytes and neutrophils | in vitro | T cells | [101] |
| IL-17A, IL-17F | high H4ac, low H3K9me3 | recruitment of monocytes and neutrophils | in vitro | CD4+ T cells | [108] |
| HPK1 | high H3K27me3 | T cell activation | in vitro | CD4+ T cells | [103] |
| CD11a/CD18 | low H3K9me3, high H3ac | leukocyte adhesion and migration in inflamed tissues | in vitro | CD4+ T cells | [78,105] |
| CD70 | low H3K9me3, high H3ac | T cell activation | in vitro | CD4+ T cells | [78,105] |
| IL-2 | low H3K18ac | Treg suppression | in vitro | T cells | [106] |
| TNFAIP3 | low H3K4me3 | production of IFNγ and IL-17 | in vitro | CD4+ T cells | [109] |
| miR-142-3p/5p | high H3K27me3, low H3K9/14ac | production of IL-21, ICOS, and CD40LG, and Tfh cell differentiation | in vitro | CD4+ T cells | [110] |
| RORγt | high H3K4me3, low H3K27me3 | Th17 differentiation | in vitro | CD4+ T cells | [111] |
| miRNA Expression | Target Genes | Effects | Types of Experiments | Cells | Refs. |
|---|---|---|---|---|---|
| miR-10b-5p upregulation | SRSF1 | Treg suppression | in vitro | T cells | [112] |
| miR-17-5p downregulation | IL-10 | autoantibody production | in vitro | B cells | [115] |
| miR-21 upregulation | PDCD4 | cell proliferation and B cell maturation | in vitro | CD4+ T cells | [116] |
| miR-21 upregulation | BDH2 | suppression of DNA methylation | in vitro | CD4+ T cells | [92] |
| miR-34a upregulation | FOXP3 | Treg suppression | in vitro | PBMCs, CD4+ T cells | [121] |
| miR-98 downregulation | FAS | apoptosis | in vitro | CD4+ T cells | [122] |
| miR-99a-3p downregulation | EIF4EBP1 | autophagy | in vitro | PBMCs | [123] |
| miR-125a downregulation | KLF13 | increase in RANTES expression | in vitro | T cells | [124] |
| miR-146a downregulation | STAT1 | Th1 responses | in vitro | PBMCs | [126,127] |
| miR-152-3p upregulation | KLF5 | increase in BAFF expression | in vitro | B cells | [129] |
| miR-152-3p upregulation | DNMT1 | autoreactive CD4+ T cell responses and TLR-mediated inflammatory responses | in vitro | PBMCs, CD4+ T cells | [130] |
| miR-155 downregulation | CREB | reduced IL-2 production | in vitro | PBMCs | [131,132] |
| miR-302d downregulation | IRF9 | type I IFN-mediated responses | in vitro | monocytes | [133] |
| miR-663 upregulation | TGF-β1 | Tfh cell activation and Treg suppression | in vitro | BMSCs | [134] |
| miR4512 downregulation | TLR | NETosis | in vitro | monocytes, macrophages | [135] |
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.J.; Karp, D.R. Autoantibodies and SLE: The threshold for disease. Nat. Rev. Rheumatol. 2014, 10, 181–186. [Google Scholar] [CrossRef]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global epidemiology of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Andersen, J.; Aringer, M.; Arnaud, L.; Bae, S.C.; Boletis, J.; Bruce, I.N.; Cervera, R.; Doria, A.; et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann. Rheum. Dis. 2024, 83, 15–29. [Google Scholar] [CrossRef]
- Wahren-Herlenius, M.; Dorner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Ellis, J.A.; Kemp, A.S.; Ponsonby, A.L. Gene-environment interaction in autoimmune disease. Expert. Rev. Mol. Med. 2014, 16, e4. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; Yuliasih, Y. Disentangling the Pathogenesis of Systemic Lupus Erythematosus: Close Ties between Immunological, Genetic and Environmental Factors. Medicina 2023, 59, 1033. [Google Scholar] [CrossRef]
- Costenbader, K.H.; Gay, S.; Alarcon-Riquelme, M.E.; Iaccarino, L.; Doria, A. Genes, epigenetic regulation and environmental factors: Which is the most relevant in developing autoimmune diseases? Autoimmun. Rev. 2012, 11, 604–609. [Google Scholar] [CrossRef]
- Wada, T.T.; Araki, Y.; Sato, K.; Aizaki, Y.; Yokota, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 2014, 444, 682–686. [Google Scholar] [CrossRef]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone Methylation and STAT-3 Differentially Regulate Interleukin-6-Induced Matrix Metalloproteinase Gene Activation in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Mimura, T. The Mechanisms Underlying Chronic Inflammation in Rheumatoid Arthritis from the Perspective of the Epigenetic Landscape. J. Immunol. Res. 2016, 2016, 6290682. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Mimura, T. The Histone Modification Code in the Pathogenesis of Autoimmune Diseases. Mediators Inflamm. 2017, 2017, 2608605. [Google Scholar] [CrossRef]
- Araki, Y.; Mimura, T. Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 905. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Aizaki, Y.; Sato, K.; Oda, H.; Kurokawa, R.; Mimura, T. Altered gene expression profiles of histone lysine methyltransferases and demethylases in rheumatoid arthritis synovial fibroblasts. Clin. Exp. Rheumatol. 2018, 36, 314–316. [Google Scholar] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef]
- Araki, Y.; Fann, M.; Wersto, R.; Weng, N.P. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: Perforin and granzyme B). J. Immunol. 2008, 180, 8102–8108. [Google Scholar] [CrossRef]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H., 3rd; Schones, D.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef]
- Araki Y: Role of histone modifications in differentiation and effector function of CD8 T cells: Update review including genome-wide analysis. Jpn. J. Clin. Immunol. 2011, 34, 131–137. [CrossRef]
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315. [Google Scholar] [CrossRef]
- Pan, L.; Lu, M.P.; Wang, J.H.; Xu, M.; Yang, S.R. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J. Pediatr. 2020, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Boulougoura, A.; Endo, Y.; Tsokos, G.C. Abnormalities of T cells in systemic lupus erythematosus: New insights in pathogenesis and therapeutic strategies. J. Autoimmun. 2022, 132, 102870. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Hsu, H.C.; Mountz, J.D. Autoreactive B cells in SLE, villains or innocent bystanders? Immunol. Rev. 2019, 292, 120–138. [Google Scholar] [CrossRef]
- Valencia, X.; Yarboro, C.; Illei, G.; Lipsky, P.E. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J. Immunol. 2007, 178, 2579–2588. [Google Scholar] [CrossRef]
- Enders, A.; Bouillet, P.; Puthalakath, H.; Xu, Y.; Tarlinton, D.M.; Strasser, A. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med. 2003, 198, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Pelanda, R.; Torres, R.M. Receptor editing for better or for worse. Curr. Opin. Immunol. 2006, 18, 184–190. [Google Scholar] [CrossRef]
- von Boehmer, H. Positive selection of lymphocytes. Cell 1994, 76, 219–228. [Google Scholar] [CrossRef]
- Starr, T.K.; Jameson, S.C.; Hogquist, K.A. Positive and negative selection of T cells. Annu. Rev. Immunol. 2003, 21, 139–176. [Google Scholar] [CrossRef]
- Cambier, J.C.; Gauld, S.B.; Merrell, K.T.; Vilen, B.J. B-cell anergy: From transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol. 2007, 7, 633–643. [Google Scholar] [CrossRef]
- Cyster, J.G.; Hartley, S.B.; Goodnow, C.C. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature 1994, 371, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D. The induction and maintenance of T cell anergy. Clin. Immunol. 2006, 120, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Van Parijs, L.; Abbas, A.K. Homeostasis and self-tolerance in the immune system: Turning lymphocytes off. Science 1998, 280, 243–248. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Ueno H: T follicular helper cells in human autoimmunity. Curr. Opin. Immunol. 2016, 43, 24–31. [CrossRef]
- Blanco P, Ueno H, Schmitt N: T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur. J. Immunol. 2016, 46, 281–290. [CrossRef]
- Marks, K.E.; Rao, D.A. T peripheral helper cells in autoimmune diseases. Immunol. Rev. 2022, 307, 191–202. [Google Scholar] [CrossRef]
- Bocharnikov, A.V.; Keegan, J.; Wacleche, V.S.; Cao, Y.; Fonseka, C.Y.; Wang, G.; Muise, E.S.; Zhang, K.X.; Arazi, A.; Keras, G.; et al. PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight 2019, 4, e130062. [Google Scholar] [CrossRef] [PubMed]
- Wehr, C.; Eibel, H.; Masilamani, M.; Illges, H.; Schlesier, M.; Peter, H.H.; Warnatz, K. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin. Immunol. 2004, 113, 161–171. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, S.; Qian, J.; Wang, Y.; Yu, X.; Dai, D.; Dai, M.; Wu, L.; Liao, Z.; Xue, Z.; et al. T-bet+CD11c+ B cells are critical for antichromatin immunoglobulin G production in the development of lupus. Arthritis Res. Ther. 2017, 19, 225. [Google Scholar] [CrossRef]
- Mouat, I.C.; Goldberg, E.; Horwitz, M.S. Age-associated B cells in autoimmune diseases. Cell. Mol. Life Sci. 2022, 79, 402. [Google Scholar] [CrossRef]
- Bolouri, N.; Akhtari, M.; Farhadi, E.; Mansouri, R.; Faezi, S.T.; Jamshidi, A.; Mahmoudi, M. Role of the innate and adaptive immune responses in the pathogenesis of systemic lupus erythematosus. Inflamm. Res Off. J. Eur. Histamine Res. Soc. 2022, 71, 537–554. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Wittmann, M.; Vital, E.M. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat. Rev. Rheumatol. 2022, 18, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A: An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783.
- Golbabapour, S.; Abdulla, M.A.; Hajrezaei, M. A concise review on epigenetic regulation: Insight into molecular mechanisms. Int. J. Mol. Sci. 2011, 12, 8661–8694. [Google Scholar] [CrossRef]
- Bassett, A.; Cooper, S.; Wu, C.; Travers, A. The folding and unfolding of eukaryotic chromatin. Curr. Opin. Genet. Dev. 2009, 19, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Bernstein, B.E. Chromatin state maps: New technologies, new insights. Curr. Opin. Genet. Dev. 2008, 18, 109–115. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Eden, S.; Cedar, H. Role of DNA methylation in the regulation of transcription. Curr. Opin. Genet. Dev. 1994, 4, 255–259. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Esteve, P.O. Mammalian DNA (cytosine-5) methyltransferases and their expression. Clin. Immunol. 2003, 109, 6–16. [Google Scholar] [CrossRef]
- Buck-Koehntop, B.A.; Defossez, P.A. On how mammalian transcription factors recognize methylated DNA. Epigenetics 2013, 8, 131–137. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yamaguchi, L.; Sharif, J.; Johmura, Y.; Kawamura, T.; Nakanishi, K.; Shimamura, S.; Arita, K.; Kodama, T.; Ishikawa, F.; et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013, 502, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, T.; Yu, X.; Xue, Z.; Shen, N. The role of long non-coding RNAs in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 657–669. [Google Scholar] [CrossRef]
- Richardson, B.; Powers, D.; Hooper, F.; Yung, R.L.; O’Rourke, K. Lymphocyte function-associated antigen 1 overexpression and T cell autoreactivity. Arthritis Rheum. 1994, 37, 1363–1372. [Google Scholar] [CrossRef]
- Yung, R.; Powers, D.; Johnson, K.; Amento, E.; Carr, D.; Laing, T.; Yang, J.; Chang, S.; Hemati, N.; Richardson, B. Mechanisms of drug-induced lupus, I.I. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J. Clin. Investig. 1996, 97, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Kaplan, M.; Ray, D.; Zacharek, S.; Gutsch, D.; Richardson, B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.; Tesselaar, K.; van Oers, M.H.; van Lier, R.A. Control of lymphocyte function through CD27-CD70 interactions. Semin. Immunol. 1998, 10, 491–499. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, A.; Richardson, B.C. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J. Immunol. 2005, 174, 6212–6219. [Google Scholar] [CrossRef] [PubMed]
- Van Kooten, C.; Banchereau, J. CD40-CD40 ligand: A multifunctional receptor-ligand pair. Adv. Immunol. 1996, 61, 1–77. [Google Scholar]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, Y.; Gao, F.; Wu, X.; Tang, J.; Yin, H.; Luo, Y.; Richardson, B.; Lu, Q. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J. Autoimmun. 2010, 35, 58–69. [Google Scholar] [CrossRef]
- Hedrich, C.M.; Rauen, T.; Apostolidis, S.A.; Grammatikos, A.P.; Rodriguez Rodriguez, N.; Ioannidis, C.; Kyttaris, V.C.; Crispin, J.C.; Tsokos, G.C. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc. Natl. Acad. Sci. USA 2014, 111, 13457–13462. [Google Scholar] [CrossRef]
- Barreto, G.; Schafer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Doderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, M.; Yin, H.; Gao, F.; Wu, X.; Luo, Y.; Zhao, S.; Zhang, X.; Su, Y.; Hu, N.; et al. Overexpression of the growth arrest and DNA damage-induced 45alpha gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum. 2010, 62, 1438–1447. [Google Scholar] [CrossRef]
- Li, Y.; Huang, C.; Zhao, M.; Liang, G.; Xiao, R.; Yung, S.; Chan, T.M.; Lu, Q. A possible role of HMGB1 in DNA demethylation in CD4+ T cells from patients with systemic lupus erythematosus. Clin. Dev. Immunol. 2013, 2013, 206298. [Google Scholar] [CrossRef] [PubMed]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, S.; Luo, S.; Wu, H.; Tang, M.; Cheng, W.; Zhang, Q.; Zhang, P.; Yu, X.; Xia, Y.; et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J. Autoimmun. 2014, 54, 127–136. [Google Scholar] [CrossRef]
- Deng, C.; Kaplan, M.J.; Yang, J.; Ray, D.; Zhang, Z.; McCune, W.J.; Hanash, S.M.; Richardson, B.C. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001, 44, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, K.; Nagpal, K.; Hedrich, C.M.; Mizui, M.; Fitzgerald, L.M.; Tsokos, G.C. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J. Biol. Chem. 2013, 288, 21936–21944. [Google Scholar] [PubMed]
- Pan, W.; Zhu, S.; Yuan, M.; Cui, H.; Wang, L.; Luo, X.; Li, J.; Zhou, H.; Tang, Y.; Shen, N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 2010, 184, 6773–6781. [Google Scholar] [CrossRef]
- Crispin, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef]
- Rauen, T.; Hedrich, C.M.; Juang, Y.T.; Tenbrock, K.; Tsokos, G.C. cAMP-responsive element modulator (CREM)alpha protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J. Biol. Chem. 2011, 286, 43437–43446. [Google Scholar] [CrossRef]
- Mok, A.; Solomon, O.; Nayak, R.R.; Coit, P.; Quach, H.L.; Nititham, J.; Sawalha, A.H.; Barcellos, L.F.; Criswell, L.A.; Chung, S.A. Genome-wide profiling identifies associations between lupus nephritis and differential methylation of genes regulating tissue hypoxia and type 1 interferon responses. Lupus. Sci. Med. 2016, 3, e000183. [Google Scholar] [CrossRef]
- Liao, W.; Li, M.; Wu, H.; Jia, S.; Zhang, N.; Dai, Y.; Zhao, M.; Lu, Q. Down-regulation of MBD4 contributes to hypomethylation and overexpression of CD70 in CD4+ T cells in systemic lupus erythematosus. Clin. Epigenetics 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, M.Y.; Gao, X.F.; Jia, S.J.; Gao, K.Q.; Zhou, Y.; Zhang, H.H.; Huang, Y.; Wang, J.; Wu, H.J.; et al. Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4+ T cells of systemic lupus erythematosus. Clin. Immunol. 2018, 187, 113–121. [Google Scholar] [CrossRef]
- Ulff-Møller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Grønbaek, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef]
- Imgenberg-Kreuz, J.; Carlsson Almlöf, J.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.L.; Rantapää-Dahlqvist, S.; Bengtsson, A.A.; Jönsen, A.; Padyukov, L.; et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef]
- Joseph, S.; George, N.I.; Green-Knox, B.; Treadwell, E.L.; Word, B.; Yim, S.; Lyn-Cook, B. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J. Autoimmun. 2019, 96, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.G.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, L.; Yang, L.; Jia, S.; Du, P.; Min, X.; Wu, J.; Wu, H.; Long, H.; Lu, Q.; et al. UHRF1 downregulation promotes T follicular helper cell differentiation by increasing BCL6 expression in SLE. Clin. Epigenetics 2021, 13, 31. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, L.; Hu, C.; Zhang, Y. Genome-wide analysis of histone H3 lysine 4 trimethylation by ChIP-chip in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Clin. Exp. Rheumatol. 2010, 28, 158–168. [Google Scholar]
- Zhou, Y.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Zhong, Q.; Zhao, M.; Lu, Q. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus 2011, 20, 1365–1371. [Google Scholar] [CrossRef]
- Sullivan, K.E.; Suriano, A.; Dietzmann, K.; Lin, J.; Goldman, D.; Petri, M.A. The TNFalpha locus is altered in monocytes from patients with systemic lupus erythematosus. Clin. Immunol. 2007, 123, 74–81. [Google Scholar] [CrossRef]
- Apostolidis, S.A.; Rauen, T.; Hedrich, C.M.; Tsokos, G.C.; Crispin, J.C. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J. Biol. Chem. 2013, 288, 26775–26784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes Immun. 2010, 11, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Long, H.; Liao, J.; Zhao, M.; Liang, G.; Wu, X.; Zhang, P.; Ding, S.; Luo, S.; Lu, Q. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J. Autoimmun. 2011, 37, 180–189. [Google Scholar] [CrossRef]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar] [PubMed]
- Zhao, M.; Wu, X.; Zhang, Q.; Luo, S.; Liang, G.; Su, Y.; Tan, Y.; Lu, Q. RFX1 regulates CD70 and CD11a expression in lupus T cells by recruiting the histone methyltransferase SUV39H1. Arthritis Res. Ther. 2010, 12, R227. [Google Scholar] [CrossRef]
- Hedrich, C.M.; Rauen, T.; Tsokos, G.C. cAMP-responsive element modulator (CREM)α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: Implications in systemic lupus erythematosus. J. Biol. Chem. 2011, 286, 43429–43436. [Google Scholar] [CrossRef]
- Mishra, N.; Brown, D.R.; Olorenshaw, I.M.; Kammer, G.M. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc. Natl. Acad. Sci. USA 2001, 98, 2628–2633. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, J.; Zhao, M.; Wu, H.; Yung, S.; Chan, T.M.; Yoshimura, A.; Lu, Q. Increased expression of TLR2 in CD4+ T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur. J. Immunol. 2015, 45, 2683–2693. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, L.; Luo, H.; Li, Q.Z.; Zuo, X. TNFAIP3 downregulation mediated by histone modification contributes to T-cell dysfunction in systemic lupus erythematosus. Rheumatology 2017, 56, 835–843. [Google Scholar] [CrossRef]
- Ding, S.; Zhang, Q.; Luo, S.; Gao, L.; Huang, J.; Lu, J.; Chen, J.; Zeng, Q.; Guo, A.; Zeng, J.; et al. BCL-6 suppresses miR-142-3p/5p expression in SLE CD4+ T cells by modulating histone methylation and acetylation of the miR-142 promoter. Cell. Mol. Immunol. 2020, 17, 474–482. [Google Scholar] [CrossRef]
- Lee, S.; Nakayamada, S.; Kubo, S.; Yamagata, K.; Yoshinari, H.; Tanaka, Y. Interleukin-23 drives expansion of Thelper 17 cells through epigenetic regulation by signal transducer and activators of transcription 3 in lupus patients. Rheumatology 2020, 59, 3058–3069. [Google Scholar] [CrossRef]
- Ramanujan, S.A.; Cravens, E.N.; Krishfield, S.M.; Kyttaris, V.C.; Moulton, V.R. Estrogen-Induced hsa-miR-10b-5p Is Elevated in T Cells From Patients With Systemic Lupus Erythematosus and Down-Regulates Serine/Arginine-Rich Splicing Factor 1. Arthritis Rheumatol. 2021, 73, 2052–2058. [Google Scholar] [CrossRef]
- Moulton, V.R.; Grammatikos, A.P.; Fitzgerald, L.M.; Tsokos, G.C. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 1845–1850. [Google Scholar] [CrossRef]
- Katsuyama, T.; Li, H.; Comte, D.; Tsokos, G.C.; Moulton, V.R. Splicing factor SRSF1 controls T cell hyperactivity and systemic autoimmunity. J. Clin. Investig. 2019, 129, 5411–5423. [Google Scholar] [CrossRef]
- Xu, L.; Wang, L.; Shi, Y.; Deng, Y.; Oates, J.C.; Kamen, D.L.; Gilkeson, G.S.; Wang, F.; Zhang, M.; Tan, W.; et al. Up-Regulated Interleukin-10 Induced by E2F Transcription Factor 2-MicroRNA-17-5p Circuitry in Extrafollicular Effector B Cells Contributes to Autoantibody Production in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2022, 74, 496–507. [Google Scholar] [CrossRef]
- Stagakis, E.; Bertsias, G.; Verginis, P.; Nakou, M.; Hatziapostolou, M.; Kritikos, H.; Iliopoulos, D.; Boumpas, D.T. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann. Rheum. Dis. 2011, 70, 1496–1506. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef]
- Hilliard, A.; Hilliard, B.; Zheng, S.J.; Sun, H.; Miwa, T.; Song, W.; Goke, R.; Chen, Y.H. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J. Immunol. 2006, 177, 8095–8102. [Google Scholar] [CrossRef]
- Bitomsky, N.; Bohm, M.; Klempnauer, K.H. Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene 2004, 23, 7484–7493. [Google Scholar] [CrossRef] [PubMed]
- Tsytsykova, A.V.; Tsitsikov, E.N.; Geha, R.S. The CD40L promoter contains nuclear factor of activated T cells-binding motifs which require AP-1 binding for activation of transcription. J. Biol. Chem. 1996, 271, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wang, J.; Gong, W.; Xu, H.; Pan, X.; Chen, Y.; Ru, S.; Wang, H.; Chen, X.; Zhao, Y.; et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J. Autoimmun. 2019, 102, 96–113. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J. Role of MiR-98 and Its Underlying Mechanisms in Systemic Lupus Erythematosus. J. Rheumatol. 2018, 45, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, B.; Deng, D. Targeting of EIF4EBP1 by miR-99a-3p affects the functions of B lymphocytes via autophagy and aggravates SLE disease progression. J. Cell Mol. Med. 2021, 25, 10291–10305. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, Y.; Qu, B.; Cui, H.; Wang, S.; Wang, L.; Luo, X.; Huang, X.; Li, J.; Chen, S.; et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 3425–3435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, J.; Qin, H.; Ge, Y.; Du, J.; Lin, J.; Zhu, X.; Wang, J.; Xu, J. Elevated expression of miR-142-3p is related to the pro-inflammatory function of monocyte-derived dendritic cells in SLE. Arthritis Res. Ther. 2016, 18, 263. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.K.; Satoh, M.; Pauley, K.M. Contrast in aberrant microRNA expression in systemic lupus erythematosus and rheumatoid arthritis: Is microRNA-146 all we need? Arthritis Rheum. 2009, 60, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef]
- Luo, S.; Ding, S.; Liao, J.; Zhang, P.; Liu, Y.; Zhao, M.; Lu, Q. Excessive miR-152-3p Results in Increased BAFF Expression in SLE B-Cells by Inhibiting the KLF5 Expression. Front. Immunol. 2019, 10, 1127. [Google Scholar] [CrossRef]
- Tao, B.; Xiang, W.; Li, X.; He, C.; Chen, L.; Xia, X.; Peng, T.; Peng, L.; Yang, X.; Zhong, C. Regulation of Toll-like receptor-mediated inflammatory response by microRNA-152-3p-mediated demethylation of MyD88 in systemic lupus erythematosus. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2021, 70, 285–296. [Google Scholar] [CrossRef]
- Miao, C.G.; Yang, Y.Y.; He, X.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.W.; Jin, Y.; Li, J. The emerging role of microRNAs in the pathogenesis of systemic lupus erythematosus. Cell Signal. 2013, 25, 1828–1836. [Google Scholar] [CrossRef]
- Lashine YA, Salah S, Aboelenein HR, Abdelaziz AI: Correcting the expression of miRNA-155 represses PP2Ac and enhances the release of IL-2 in PBMCs of juvenile SLE patients. Lupus 2015, 24, 240–247. [CrossRef]
- Smith, S.; Fernando, T.; Wu, P.W.; Seo, J.; Gabhann, J.N.; Piskareva, O.; McCarthy, E.; Howard, D.; O’Connell, P.; Conway, R.; et al. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. J. Autoimmun. 2017, 79, 105–111. [Google Scholar] [CrossRef]
- Geng, L.; Tang, X.; Zhou, K.; Wang, D.; Wang, S.; Yao, G.; Chen, W.; Gao, X.; Chen, W.; Shi, S.; et al. MicroRNA-663 induces immune dysregulation by inhibiting TGF-β1 production in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Cell. Mol. Immunol. 2019, 16, 260–274. [Google Scholar] [CrossRef]
- Yang, B.; Huang, X.; Xu, S.; Li, L.; Wu, W.; Dai, Y.; Ge, M.X.; Yuan, L.; Cao, W.; Yang, M.; et al. Decreased miR-4512 Levels in Monocytes and Macrophages of Individuals With Systemic Lupus Erythematosus Contribute to Innate Immune Activation and Neutrsophil NETosis by Targeting TLR4 and CXCL2. Front. Immunol. 2021, 12, 756825. [Google Scholar] [CrossRef]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2023. [Google Scholar] [CrossRef]
- Liu, Q.; Deng, Y.; Li, C.; Xie, H.; Liu, Q.; Ming, S.; Wu, D.; Luo, F. LncRNA GAS5 suppresses CD4+ T cell activation by upregulating E4BP4 via inhibiting miR-92a-3p in systemic lupus erythematosus. Immunol. Lett. 2020, 227, 41–47. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, L.; Qian, J.; Qu, B.; Xia, S.; La, T.; Wu, Y.; Ma, J.; Zeng, J.; Guo, Q.; et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J. Autoimmun. 2016, 75, 96–104. [Google Scholar] [CrossRef]
- Gao, F.; Tan, Y.; Luo, H. MALAT1 is involved in type I IFNs-mediated systemic lupus erythematosus by up-regulating OAS2, OAS3, and OASL. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Medicas E Biol. 2020, 53, e9292. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araki, Y.; Mimura, T. Epigenetic Dysregulation in the Pathogenesis of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2024, 25, 1019. https://doi.org/10.3390/ijms25021019
Araki Y, Mimura T. Epigenetic Dysregulation in the Pathogenesis of Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2024; 25(2):1019. https://doi.org/10.3390/ijms25021019
Chicago/Turabian StyleAraki, Yasuto, and Toshihide Mimura. 2024. "Epigenetic Dysregulation in the Pathogenesis of Systemic Lupus Erythematosus" International Journal of Molecular Sciences 25, no. 2: 1019. https://doi.org/10.3390/ijms25021019