
Activation of Young LINE-1 Elements by CRISPRa

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
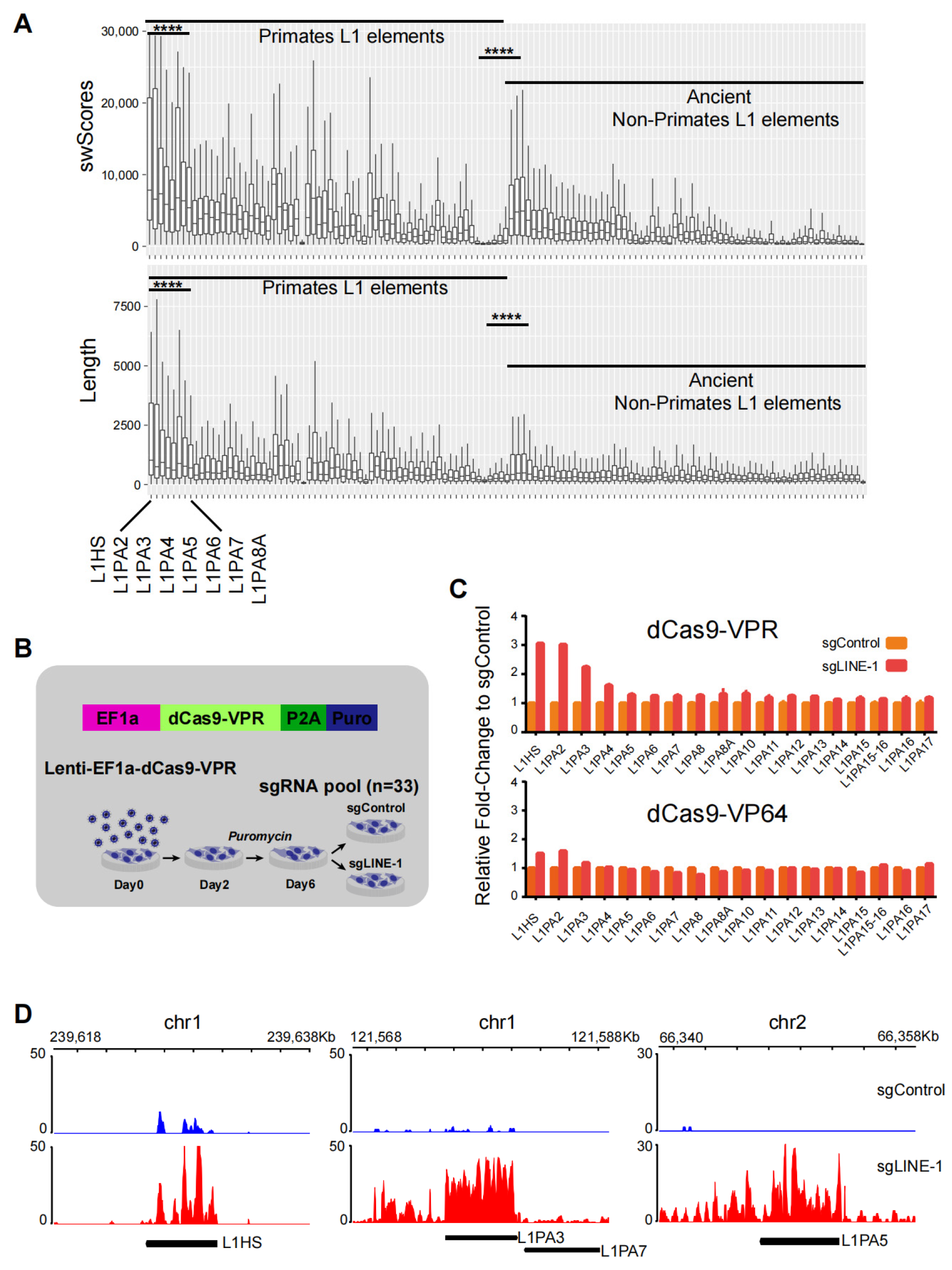
2.1. Efficient Activation of Young LINE-1 by CRISPRa
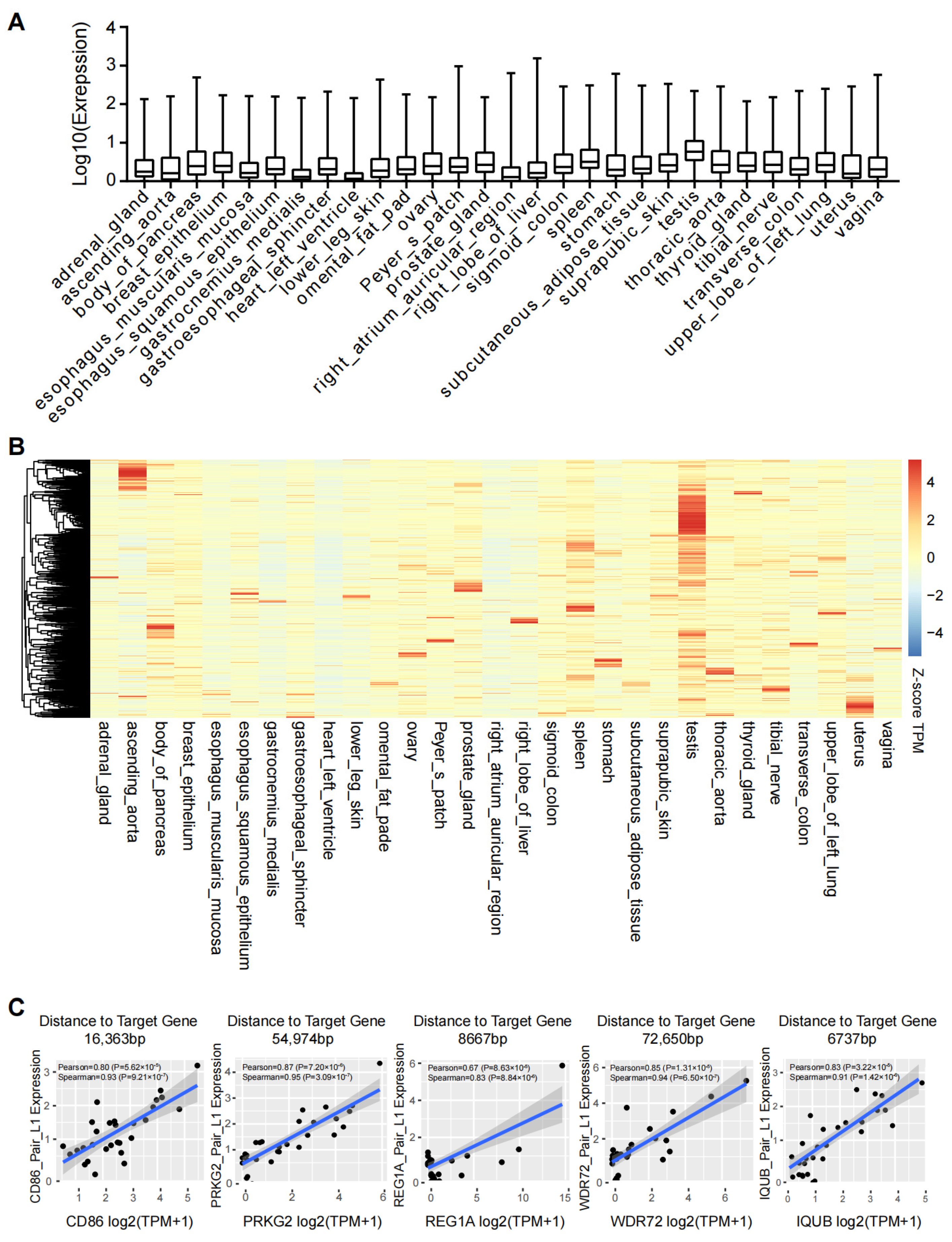
2.2. Activation of nearby Genes by LINE-1 Elements

2.3. Modulation of nearby Genes by Sense and Anti-Sense Transcripts of L1s
2.4. Identification of Differentially Expressed Genes by L1 Activation
2.5. L1s Are Expressed in a Human Tissue Specific Manner
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids for the CRISPRa System
4.3. sgRNA Design
4.4. Lentiviral Production in HEK293T Cells
4.5. LINE-1 Activation in L02 Cell Lines
4.6. Next-Generation Sequencing and Bioinformatics
4.7. ChIP-Seq Analysis
4.8. Gene Ontology Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Short | Full Name |
| ASAH2 | Neutral ceramidase |
| CTCF | CCCTC-binding factor |
| ChIP-seq | Chromatin Immunoprecipitation-sequencing |
| CRISPR | clustered regularly interspaced short palindromic repeat |
| CRISPRa | CRISPR activation |
| dCas9 | dead CRISPR-associated endonuclease Cas9 |
| DEGs | differentially expressed genes |
| DNMT1 | DNA (cytosine-5)-methyltransferase 1 |
| EMBP1 | DNA-binding protein |
| ERVs | endogenous retrovirus |
| ESC | Embryonic stem cell |
| ENCODE | The Encylopedia of DNA Elements |
| FL-L1s | full-length L1s |
| gRNAs | guide RNAs |
| GO | gene ontology |
| HUSH | The human silencing hub complex |
| hg38 | human genome 38 |
| KAP1 | cAMP-dependent protein kinase type I-beta regulatory subunit |
| KEGG | Kyoto Gene and Genomic Encyclopedia |
| L1M | L1 mammalian-specific |
| L1P | L1 primate-specific |
| LINE-1 | Long interspersed element-1 |
| L1HS/L1PA1 | L1 human specific |
| Morc2 | ATPase MORC2 |
| MOI | Multiplicity of infection |
| Mecp2 | methyl-CpG binding protein 2 |
| POGZ | Pogo transposable element with ZNF domain |
| REs | repetitive elements |
| REG1A | Lithostathine-1-alpha |
| RNA-seq | RNA-sequencing |
| RPKM | Reads Per Kilobase per Million |
| swScore | Smith-Waterman Score |
| SETDB1 | Histone-lysine N-methyltransferase SETDB1 |
| SINE | short interspersed element |
| TPM | Transcript per million |
| UTR | Untranslated Region |
| UCSC | University of California Santa Cruz |
| VP64 | The tetramer of virus protein 16 |
| VPR | VP64, P65, and Rta |
| WDR72 | WD repeat-containing protein 72 |
| YY1 | Transcriptional repressor protein YY1 |
| ZGA | Zygotic genome activation |
References
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 Retrotransposition Activity in Human Genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Briggs, E.M.; Mita, P.; Sun, X.J.; Ha, S.; Vasilyev, N.; Leopold, Z.R.; Nudler, E.; Boeke, J.D.; Logan, S.K. Unbiased proteomic mapping of the LINE-1 promoter using CRISPR Cas9. Mobile DNA 2021, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Saeliw, T.; Permpoon, T.; Iadsee, N.; Tencomnao, T.; Hu, V.W.; Sarachana, T.; Green, D.; Sae-Lee, C. LINE-1 and methylation signatures in autism spectrum disorder and their associations with the expression of autism-related genes. Sci. Rep. 2022, 12, 13970. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, M.; Li, L.; Zhou, J.S.; Lang, B.; Wang, X.P. L1 Retrotransposons: A Potential Endogenous Regulator for Schizophrenia. Front. Genet. 2022, 13, 878508. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lin, C.J.; Yin, Y.F.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.H.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e19. [Google Scholar] [CrossRef] [PubMed]
- Attig, J.; Agostini, F.; Gooding, C.; Chakrabarti, A.M.; Singh, A.; Haberman, N.; Zagalak, J.A.; Emmett, W.; Smith, C.W.J.; Luscombe, N.M.; et al. Heteromeric RNP Assembly at LINEs Controls Lineage-Specific RNA Processing. Cell 2018, 174, 1067–1081.e17. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, W.Q.; Guo, J.Y.; Liu, Y.Y.; Liu, X.L.; Liu, J.; Dou, X.Y.; Le, R.R.; Huang, Y.X.; Li, C.; et al. Nuclear m6A reader YTHDC1 regulates the scaffold function of LINE1 RNA in mouse ESCs and early embryos. Protein Cell 2021, 12, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, D.; Simi, A.; Lau, P.; Armaos, A.; Ansaloni, F.; Codino, A.; Damiani, D.; Floreani, L.; Di Carlo, V.; Vozzi, D.; et al. LINE-1 regulates cortical development by acting as long non-coding RNAs. Nat. Commun. 2023, 14, 4974. [Google Scholar] [CrossRef]
- Marasca, F.; Sinha, S.; Vadalà, R.; Polimeni, B.; Ranzani, V.; Paraboschi, E.M.; Burattin, F.V.; Ghilotti, M.; Crosti, M.; Negri, M.L.; et al. LINE1 are spliced in non-canonical transcript variants to regulate T cell quiescence and exhaustion. Nat Genet. 2022, 54, 180–193. [Google Scholar] [CrossRef]
- Jönsson, M.E.; Brattås, P.L.; Gustafsson, C.; Petri, R.; Yudovich, D.; Pircs, K.; Verschuere, S.; Madsen, S.; Hansson, J.; Larsson, J.; et al. Activation of neuronal genes via LINE-1 elements upon global DNA demethylation in human neural progenitors. Nat. Commun. 2019, 10, 3182. [Google Scholar] [CrossRef]
- Maze, I.; Feng, J.A.; Wilkinson, M.B.; Sun, H.S.; Shen, L.; Nestler, E.J. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc. Natl. Acad. Sci. USA 2011, 108, 3035–3040. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Wang, R.Y.; Lee, J.H.; Li, S.L.; Chen, S.F.; Liao, Z.; Al Hasani, L.; Nguyen, P.T.; Zhu, X.Y.; Krakowiak, J.; et al. RNA m6A modification orchestrates a LINE-1-host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 2021, 31, 861–885. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Liu, Z.P.; Wu, Z.M.; Ren, J.; Fan, Y.L.; Sun, L.; Cao, G.; Niu, Y.Y.; Zhang, B.H.; Ji, Q.Z.; et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell 2023, 186, 287–304.e26. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Zhang, T.Z.; Tong, B.; Cheng, L.X.; Jiang, W.; Sun, Y.H. POGZ suppresses 2C transcriptional program and retrotransposable elements. Cell Rep. 2023, 42, 112867. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.A.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kalia, V.; Perera, F.; Herbstman, J.; Li, T.; Nie, J.; Qu, L.R.; Yu, J.; Tang, D. Prenatal airborne polycyclic aromatic hydrocarbon exposure, LINE1 methylation and child development in a Chinese cohort. Environ. Int. 2017, 99, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Zhou, X.; Li, Y.; Zhang, O.; Zhou, Z.; Li, T.; Yuan, W.; Li, R.; Li, D.K. LINE-1 hypomethylation in spermatozoa is associated with Bisphenol A exposure. Andrology 2014, 2, 138–144. [Google Scholar] [CrossRef]
- Takahashi, T.; Stoiljkovic, M.; Song, E.; Gao, X.B.; Yasumoto, Y.; Kudo, E.; Carvalho, F.; Kong, Y.; Park, A.; Shanabrough, M.; et al. LINE-1 activation in the cerebellum drives ataxia. Neuron 2022, 110, 3278–3287.e8. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.R.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Athanikar, J.N.; Badge, R.M.; Moran, J.V. A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 2004, 32, 3846–3855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Wu, X.N.; Shi, T.T.; Xu, H.T.; Yi, J.; Shen, H.F.; Huang, M.F.; Shu, X.Y.; Wang, F.F.; Peng, B.L.; et al. Regulation of Transcription Factor Yin Yang 1 by SET7/9-mediated Lysine Methylation. Sci. Rep. 2016, 6, 21718. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Delisle, L.; Rabbani, L.; Wolff, J.; Bhardwaj, V.; Backofen, R.; Gruning, B.; Ramirez, F.; Manke, T. pyGenomeTracks: Reproducible plots for multivariate genomic datasets. Bioinformatics 2021, 37, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed]
- Roller, M.; Stamper, E.; Villar, D.; Izuogu, O.; Martin, F.; Redmond, A.M.; Ramachanderan, R.; Harewood, L.; Odom, D.T.; Flicek, P. LINE retrotransposons characterize mammalian tissue-specific and evolutionarily dynamic regulatory regions. Genome Biol. 2021, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Kotake, Y.; Asada, R.; Saunders, D.; Broyles, R.H.; Towner, R.A.; Fukui, H.; Floyd, R.A. LINE-1 hypomethylation in a choline-deficiency-induced liver cancer in rats: Dependence on feeding period. J. Biomed. Biotechnol. 2006, 2006, 17142. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Duan, S.; Hua, X.; Xu, X.; Li, Y.; Menolfi, D.; Zhou, H.; Lu, C.; Zha, S.; Goff, S.P.; et al. Asymmetric distribution of parental H3K9me3 in S phase silences L1 elements. Nature 2023, 623, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, M.; He, J.; Wu, K.; Lin, S.; Jin, L.; Chen, Y.; Liu, H.; Shi, J.; Wang, X.; et al. The RNA m(6)A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 2021, 591, 322–326. [Google Scholar] [CrossRef]
- Dai, Z.Y.; Li, R.; Hou, Y.Y.; Li, Q.; Zhao, K.; Li, T.; Li, M.J.; Wu, X.D. Inducible CRISPRa screen identifies putative enhancers. J. Genet. Genom. 2021, 48, 917–927. [Google Scholar] [CrossRef]
- Sun, X.; Peng, X.; Cao, Y.; Zhou, Y.; Sun, Y. ADNP promotes neural differentiation by modulating Wnt/beta-catenin signaling. Nat. Commun. 2020, 11, 2984. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, B.; Sun, Y. Activation of Young LINE-1 Elements by CRISPRa. Int. J. Mol. Sci. 2024, 25, 424. https://doi.org/10.3390/ijms25010424
Tong B, Sun Y. Activation of Young LINE-1 Elements by CRISPRa. International Journal of Molecular Sciences. 2024; 25(1):424. https://doi.org/10.3390/ijms25010424
Chicago/Turabian StyleTong, Bei, and Yuhua Sun. 2024. "Activation of Young LINE-1 Elements by CRISPRa" International Journal of Molecular Sciences 25, no. 1: 424. https://doi.org/10.3390/ijms25010424