
Peptidomics Unveils Distinct Acetylation Patterns of Histone and Annexin A1 in Differentiated Thyroid Cancer

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
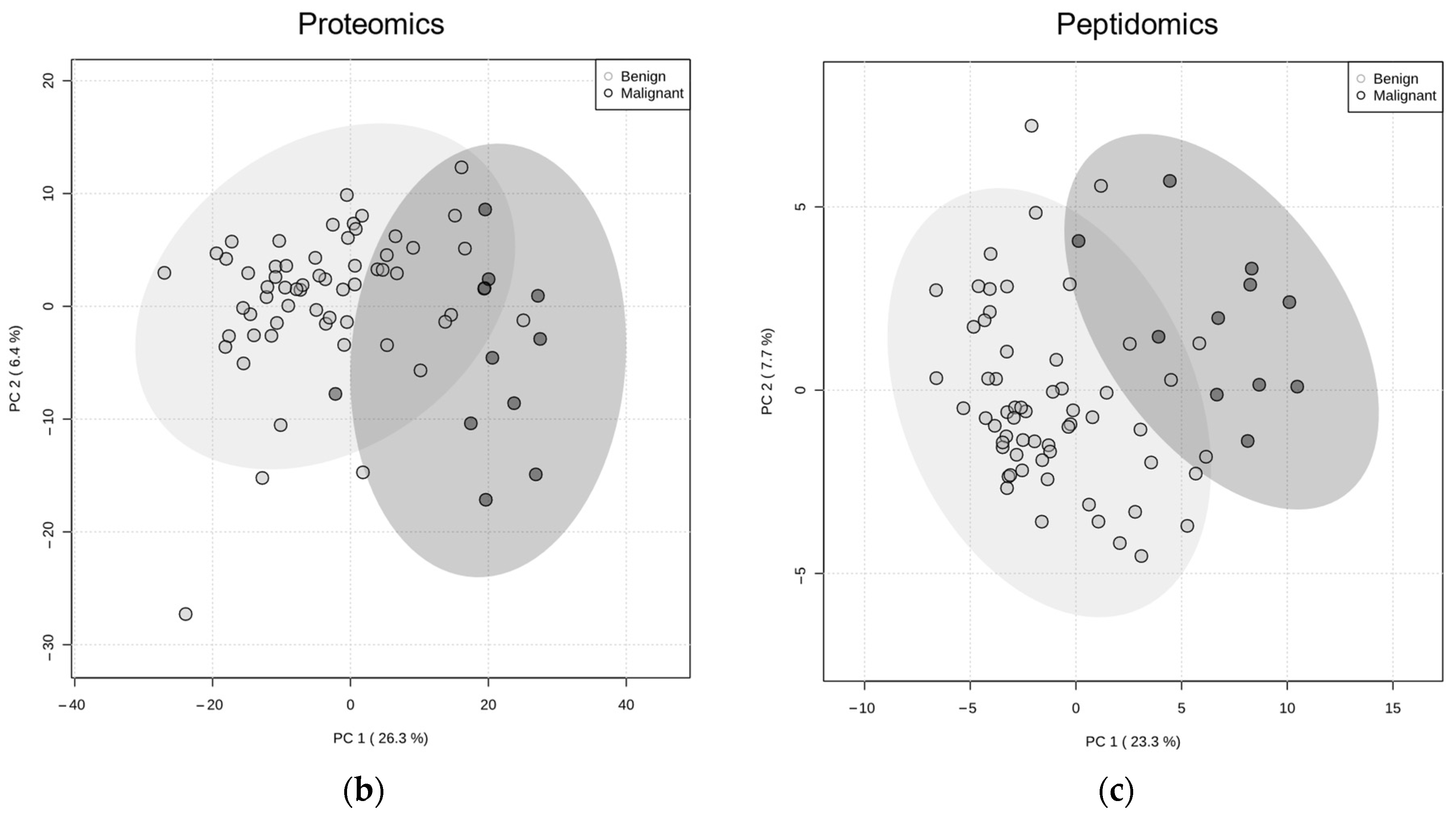
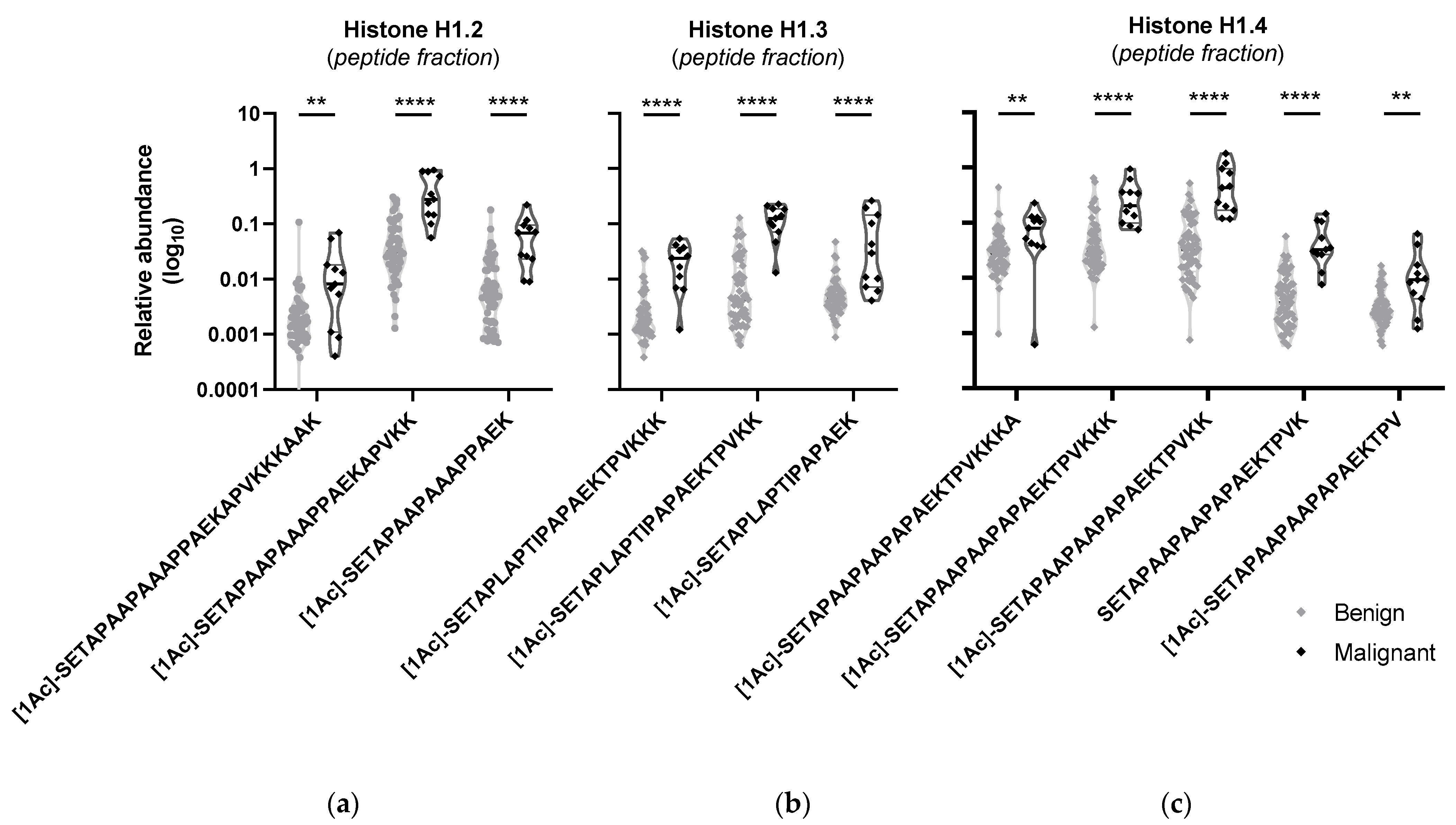
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
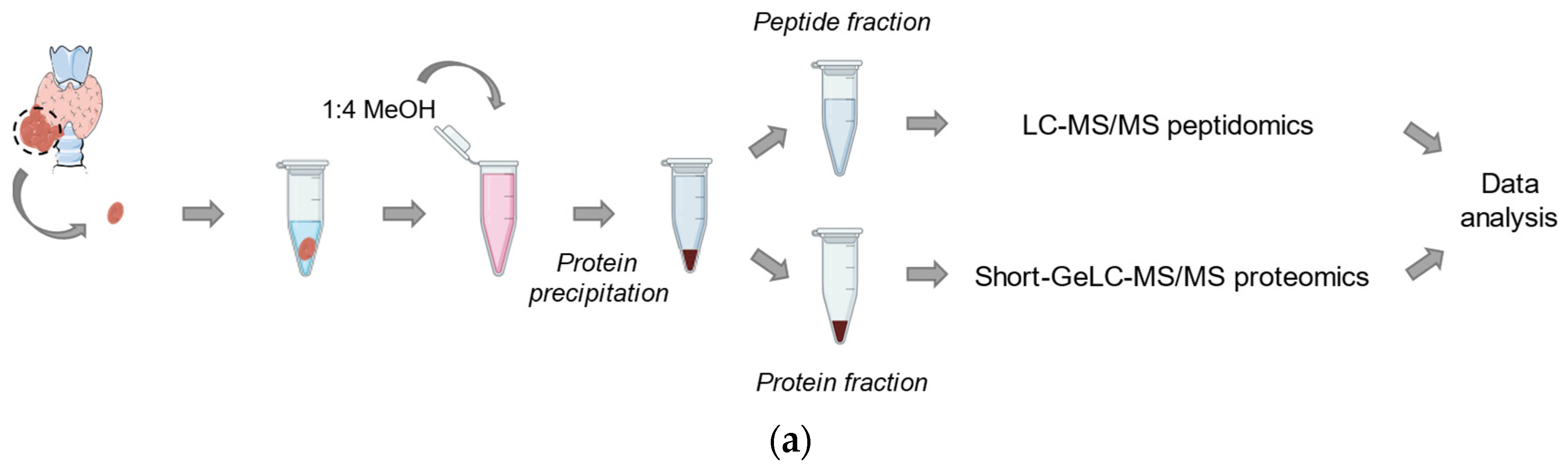
4.2. Peptidomics
4.3. Proteomics
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Grussendorf, M.; Ruschenburg, I.; Brabant, G. Malignancy rates in thyroid nodules: A long-term cohort study of 17,592 patients. Eur. Thyroid. J. 2022, 11, e220027. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.Z.; Baloch, Z.W.; Cochand-Priollet, B.; Schmitt, F.C.; Vielh, P.; VanderLaan, P.A. The 2023 Bethesda System for Reporting Thyroid Cytopathology. Thyroid 2023, 33, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Damante, G.; Scaloni, A.; Tell, G. Thyroid tumors: Novel insights from proteomic studies. Expert Rev. Proteom. 2009, 6, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Pagni, F.; L’Imperio, V.; Bono, F.; Garancini, M.; Roversi, G.; De Sio, G.; Galli, M.; Smith, A.J.; Chinello, C.; Magni, F. Proteome analysis in thyroid pathology. Expert Rev. Proteom. 2015, 12, 375–390. [Google Scholar] [CrossRef]
- Foreman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A Review of Clinical Applications and Methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef]
- Schulz-Knappe, P.; Zucht, H.D.; Heine, G.; Jurgens, M.; Hess, R.; Schrader, M. Peptidomics: The comprehensive analysis of peptides in complex biological mixtures. Comb. Chem. High. Throughput Screen. 2001, 4, 207–217. [Google Scholar] [CrossRef]
- Coelho, M.; Capela, J.; Anjo, S.I.; Pacheco, J.; Fernandes, M.S.; Amendoeira, I.; Jones, J.G.; Raposo, L.; Manadas, B. Proteomics Reveals mRNA Regulation and the Action of Annexins in Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 14542. [Google Scholar] [CrossRef]
- Krause, K.; Jessnitzer, B.; Fuhrer, D. Proteomics in thyroid tumor research. J. Clin. Endocrinol. Metab. 2009, 94, 2717–2724. [Google Scholar] [CrossRef]
- Ucal, Y.; Ozpinar, A. Proteomics in thyroid cancer and other thyroid-related diseases: A review of the literature. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140510. [Google Scholar] [CrossRef]
- Liu, X.; Yang, M.; Guo, Y.; Lu, X. Annexin A10 is a novel prognostic biomarker of papillary thyroid cancer. Ir. J. Med. Sci. 2021, 190, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; Festa, M.; Ercolino, S.F.; Zerilli, M.; Stassi, G.; Solito, E.; Parente, L. Annexin-1 downregulation in thyroid cancer correlates to the degree of tumor differentiation. Cancer Biol. Ther. 2006, 5, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ma, W.; Li, X.; Li, H.; Li, J.; Li, H.; He, F. ANXA1 enhances tumor proliferation and migration by regulating epithelial-mesenchymal transition and IL-6/JAK2/STAT3 pathway in papillary thyroid carcinoma. J. Cancer 2021, 12, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qiu, T.; Wei, G.; Que, Y.; Wang, W.; Kong, Y.; Xie, T.; Chen, X. Role of Histone Post-Translational Modifications in Inflammatory Diseases. Front. Immunol. 2022, 13, 852272. [Google Scholar] [CrossRef]
- Puppin, C.; Passon, N.; Lavarone, E.; Di Loreto, C.; Frasca, F.; Vella, V.; Vigneri, R.; Damante, G. Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 2011, 411, 679–683. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, D.; Liu, Q.; Luo, Y.; Tian, Y.; Xiao, X.; Sang, Y.; Liu, Y.; Hong, S.; Yu, S.; et al. Genome-Wide Histone H3K27 Acetylation Profiling Identified Genes Correlated With Prognosis in Papillary Thyroid Carcinoma. Front. Cell Dev. Biol. 2021, 9, 682561. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef]
- Hays, M.T. Thyroid hormone and the gut. Endocr. Res. 1988, 14, 203–224. [Google Scholar] [CrossRef]
- Niu, K.; Guo, C.; Teng, S.; Zhou, D.; Yu, S.; Yin, W.; Wang, P.; Zhu, W.; Duan, M. Pepsin promotes laryngopharyngeal neoplasia by modulating signaling pathways to induce cell proliferation. PLoS ONE 2020, 15, e0227408. [Google Scholar] [CrossRef]
- Sereg-Bahar, M.; Jerin, A.; Hocevar-Boltezar, I. Higher levels of total pepsin and bile acids in the saliva as a possible risk factor for early laryngeal cancer. Radiol. Oncol. 2015, 49, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Samuels, T.L.; Zimmermann, M.T.; Zeighami, A.; Demos, W.; Southwood, J.E.; Blumin, J.H.; Bock, J.M.; Johnston, N. RNA Sequencing Reveals Cancer-Associated Changes in Laryngeal Cells Exposed to Non-Acid Pepsin. Laryngoscope 2021, 131, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Sloane, B.F. Cathepsin B: Multiple roles in cancer. Proteom. Clin. Appl. 2014, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Song, M.J.; Jang, H.H.; Chung, Y.S. Clinicopathologic Analysis of Cathepsin B as a Prognostic Marker of Thyroid Cancer. Int. J. Mol. Sci. 2020, 21, 9537. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, T.; Nishida, S.; Nakano, T.; Funasaka, K.; Kimoto, S.; Murata, K.; Tomura, T. Study on cathepsin B activity in human thyroid tumors. Auris Nasus Larynx 1995, 22, 43–48. [Google Scholar] [CrossRef]
- Zhang, X.; Di, C.; Chen, Y.; Wang, J.; Su, R.; Huang, G.; Xu, C.; Chen, X.; Long, F.; Yang, H.; et al. Multilevel regulation and molecular mechanism of poly (rC)-binding protein 1 in cancer. FASEB J. 2020, 34, 15647–15658. [Google Scholar] [CrossRef]
- Anjo, S.I.; Simoes, I.; Castanheira, P.; Graos, M.; Manadas, B. Use of recombinant proteins as a simple and robust normalization method for untargeted proteomics screening: Exhaustive performance assessment. Talanta 2019, 205, 120163. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]
- Anjo, S.I.; Santa, C.; Manadas, B. Short GeLC-SWATH: A fast and reliable quantitative approach for proteomic screenings. Proteomics 2015, 15, 757–762. [Google Scholar] [CrossRef]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.e.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Fortelny, N.; Yang, S.; Pavlidis, P.; Lange, P.F.; Overall, C.M. Proteome TopFIND 3.0 with TopFINDer and PathFINDer: Database and analysis tools for the association of protein termini to pre- and post-translational events. Nucleic Acids Res. 2015, 43, D290–D297. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Lloyd, R.; Osamura, R.; Rosai, J. WHO Classification of Tumours Editorial Board. Endocrine and Neuroendocrine Tumours; IARC: Lyon, France, 2022. [Google Scholar]
- Tang, W.H.; Shilov, I.V.; Seymour, S.L. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J. Proteome Res. 2008, 7, 3661–3667. [Google Scholar] [CrossRef]
- Sennels, L.; Bukowski-Wills, J.C.; Rappsilber, J. Improved results in proteomics by use of local and peptide-class specific false discovery rates. BMC Bioinform. 2009, 10, 179. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sex | Female (%) | Age (Years) | BMI (kg/m2) | Free T4 (ng/dL) | TSH (um/L) |
|---|---|---|---|---|---|---|
| Benign | Female = 24 Male = 9 | 72.7 | 58.4 ± 2.3 | 28.7 ± 0.9 ** | 1.11 ± 0.07 | 0.83 ± 0.12 |
| Malignant | Female = 8 Male = 2 | 80.0 | 53.2 ± 7.1 | 23.7 ± 1.0 | 1.08 ± 0.04 | 1.28 ± 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, M.; Capela, J.; Mendes, V.M.; Pacheco, J.; Fernandes, M.S.; Amendoeira, I.; Jones, J.G.; Raposo, L.; Manadas, B. Peptidomics Unveils Distinct Acetylation Patterns of Histone and Annexin A1 in Differentiated Thyroid Cancer. Int. J. Mol. Sci. 2024, 25, 376. https://doi.org/10.3390/ijms25010376
Coelho M, Capela J, Mendes VM, Pacheco J, Fernandes MS, Amendoeira I, Jones JG, Raposo L, Manadas B. Peptidomics Unveils Distinct Acetylation Patterns of Histone and Annexin A1 in Differentiated Thyroid Cancer. International Journal of Molecular Sciences. 2024; 25(1):376. https://doi.org/10.3390/ijms25010376
Chicago/Turabian StyleCoelho, Margarida, João Capela, Vera M. Mendes, João Pacheco, Margarida Sá Fernandes, Isabel Amendoeira, John G. Jones, Luís Raposo, and Bruno Manadas. 2024. "Peptidomics Unveils Distinct Acetylation Patterns of Histone and Annexin A1 in Differentiated Thyroid Cancer" International Journal of Molecular Sciences 25, no. 1: 376. https://doi.org/10.3390/ijms25010376