
Portrayal of NLRP3 Inflammasome in Atherosclerosis: Current Knowledge and Therapeutic Targets

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
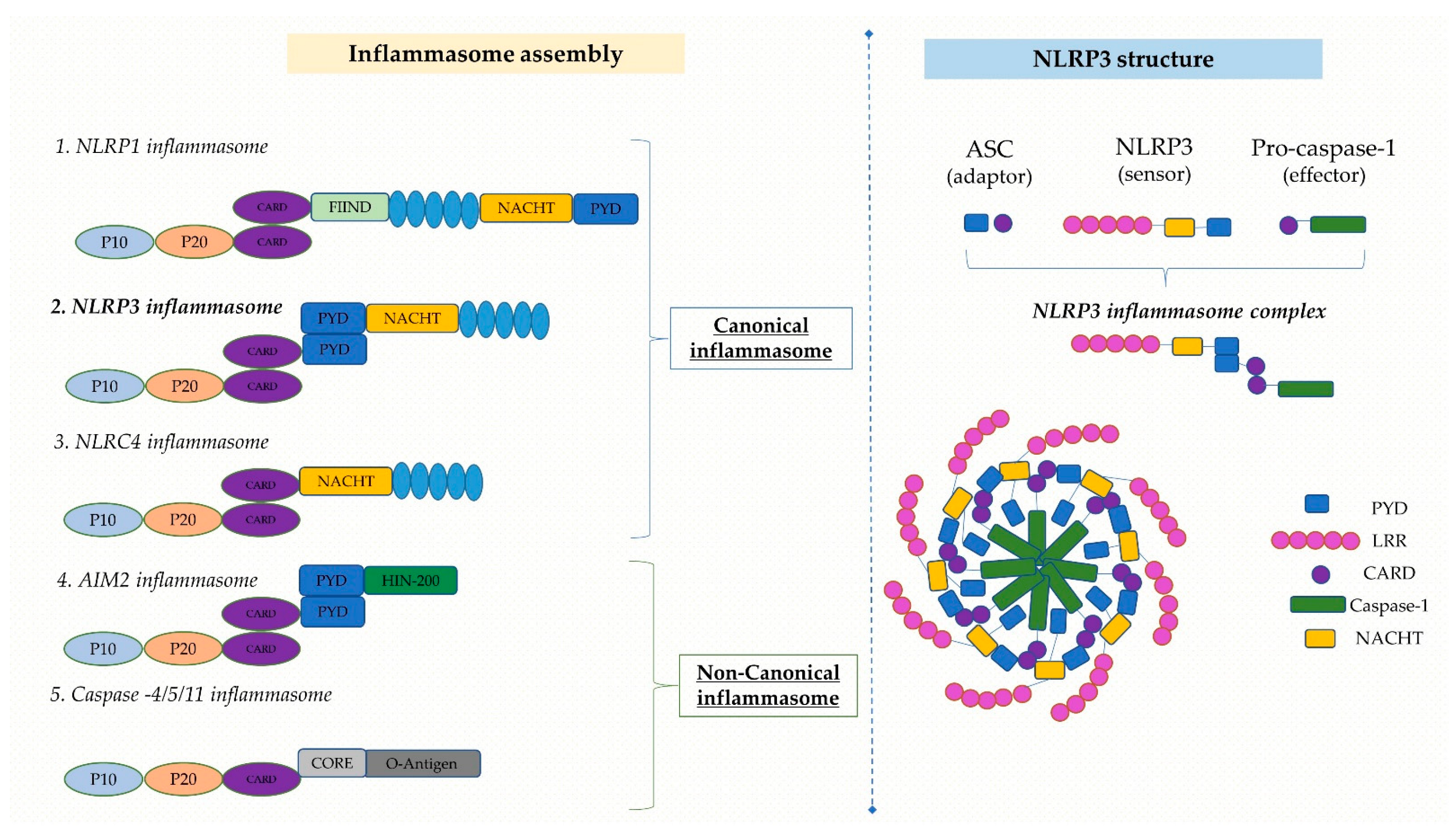
2. Portrayal of NLRP3 Inflammasome
2.1. Structure of NLRP3 Inflammasome
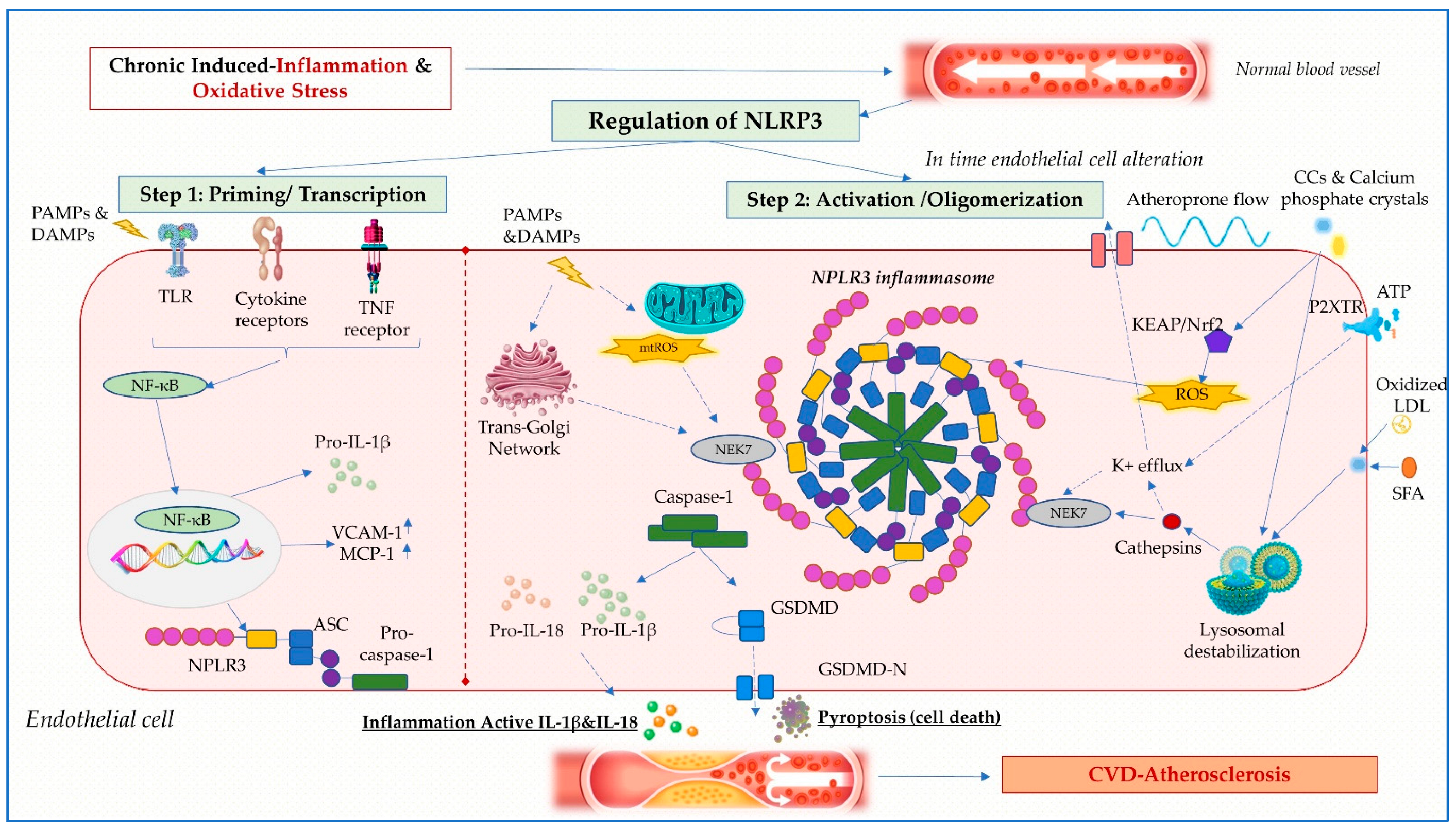
2.2. Mechanisms of NLRP3 Inflammasome Activation
2.2.1. Ionic Fluxes
2.2.2. Oxidative Stress
2.2.3. Lysosomal Damage, Autophagy and the Trans-Golgi Network
2.2.4. Regulation of NLRP3 Inflammasome
2.3. Role of NLRP3 Inflammasome in Atherosclerosis

{kind=link}
{kind=link}
{kind=link}
| Subjects | Notable NLRP3 Effects in ATS | Refs. |
|---|---|---|
| Ascending aortic tissue (CABG patients) | - NLRP3 expression higher in patients with AS and correlated with the degree of coronary artery disease | [66] |
| Human carotid atherosclerotic plaques | - NLRP3 inflammasome and components (ASC, caspase-1, IL-1β and IL-18) higher expression in unstable atherosclerotic plaques | [67] |
| Atherosclerotic plaques (ischemic cerebrovascular disease, MI patients) | - NLRP3–mRNA expression higher in symptomatic AS patients | [68] |
| Peripheral blood monocyte (chronic heart disease and acute coronary syndrome patients) | - NLRP3 inflammasome positive correlation with coronary atherosclerosis | [69] |
| ApoE−/− mice fed with a HF and HM diet | - Increased NLRP3 expression and proinflammatory effect in hyperhomocysteinemia-induced atherosclerosis | [70] |
| ApoE−/− mice fed with a HF diet | - NLRP3 inflammasome inhibition increased plaque stability | [71] |
| - NLRP3 inflammasome inhibition reduced the size of atherosclerotic plaques and IL-1β and IL-18 levels | [72] | |
| - CCs activate NLRP3 inflammasome | [52] | |
| ApoE−/− mice chow diet | - NLRP3 inflammasome activation via Sirt3/FOXO3a/Parkin signaling pathway reduced atherosclerotic progression | [73] |
| ApoE−/− mice western-type diet | - Specific NLRP3 inflammasome inhibition reduced atherosclerotic plaque development | [74] |
| Ldlr−/− mice fed with PUFAs diet | - NLRP3 inflammasome inhibition reduced atherosclerosis by macrophage autophagy activation | [75] |
| ApoE−/− mice | - The oxLDLs promote direct NLRP3 inflammasome activation and indirect via ERK1/2 pathway | [76] |
| ApoE−/−/caspase-1−/− double knockout mice | - The extent of the area of atherosclerotic plaque reduced in caspase-1 deficient mice | [77] |
| Macrophages incubated with oxLDLs | - NLRP3 inflammasome activation, increase in IL-1β and IL-18 levels | [52] |
| NLRP3-deficient THP-1 cells | - NLRP3 inhibition reduces foam cell formation of THP-1 macrophages by oxLDL uptake suppression and increasing cholesterol efflux | [73] |
| HAECs | - NLRP3 inflammasome is activated by nicotine which promotes pyroptosis, proinflammatory cytokines secretion and atherosclerosis | [78] |
| Human and mice aortic endothelial cells | - Melatonin inhibits pyroptosis through the MEG3/miR-223/ NLRP3 signaling axis | [79] |
| HAECs | - NLRP3 inhibitor Microrna-30c-5p inhibits inflammation and pyroptosis via F0X03 pathway | [80] |
| VSMC | - AIM2 can stimulate caspase-1 via NLRP3 pathway and then mediates the inflammatory response by slicing GSDMD | [81] |
3. Atherosclerosis
3.1. Endothelial Dysfunction and Oxidative Stress
3.2. Inflammation in Atherosclerosis
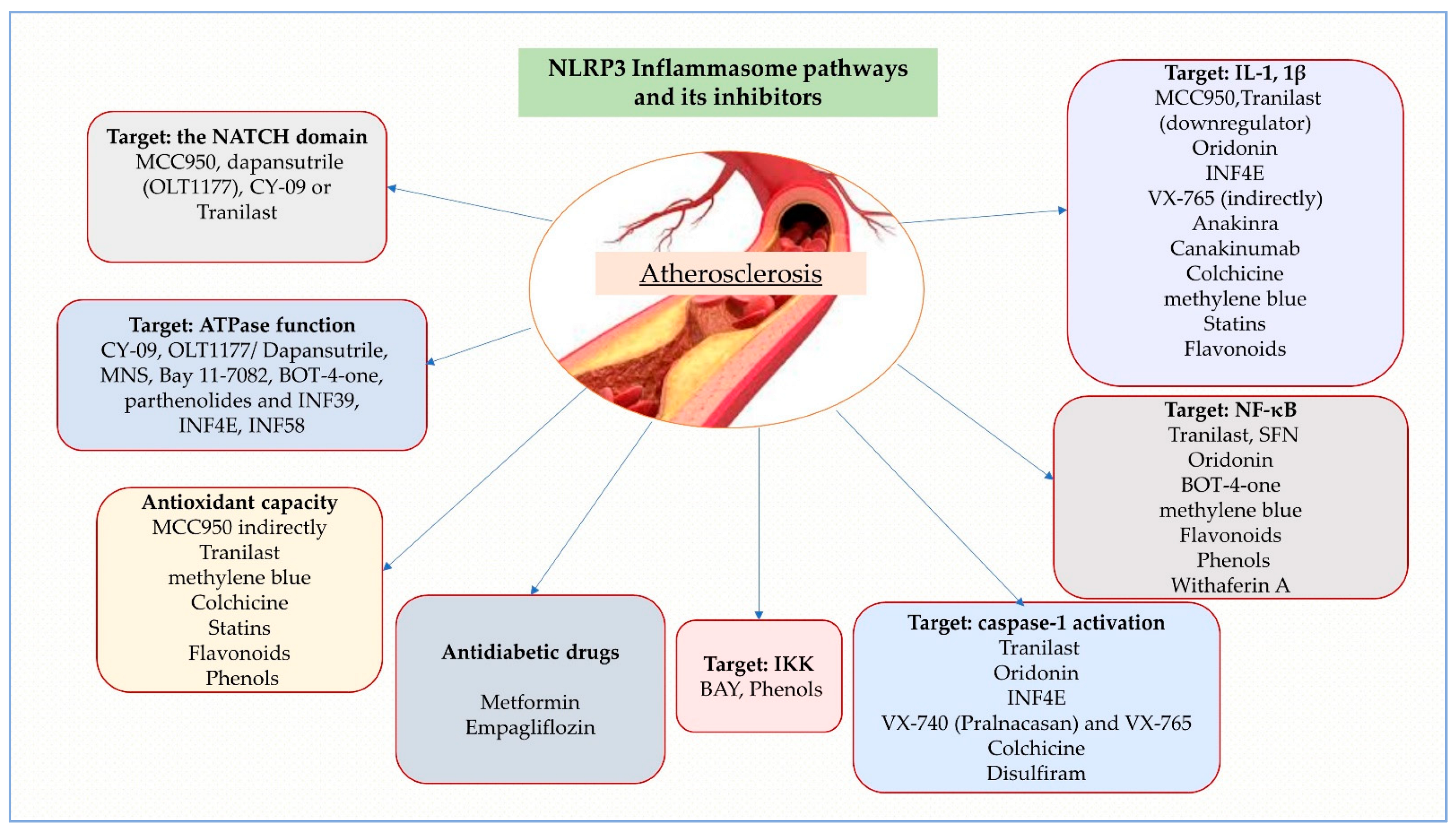
4. Therapeutic Targets
4.1. Hypoglycemic Agents
4.2. Direct and Indirect NLRP3 Inhibitors
4.2.1. MCC950
4.2.2. CY-09
4.2.3. OLT1177/Dapansutrile
4.2.4. Tranilast
4.2.5. Oridonin
4.2.6. 3,4-Methylenedioxy-β-Nitrostyrene (MNS)
4.2.7. INF Analogues
4.2.8. BAY 11-7082
4.2.9. VX-740 (Pralnacasan) and VX-765
4.2.10. Anakinra (Kineret), Rilonacept (Arcalyst) and Canakinumab (Ilaris)
4.2.11. Colchicine
4.2.12. Less Known NLRP3 Inhibitors
4.3. Statins
4.4. Natural Compounds
4.4.1. Flavonoids
4.4.2. Phenols
4.4.3. Miscellaneous
5. Discussion

6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Liberale, L.; Montecucco, F.; Schwarz, L.; Lüscher, T.F.; Camici, G.G. Inflammation and Cardiovascular Diseases: Lessons from Seminal Clinical Trials. Cardiovasc. Res. 2021, 117, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Liberale, L.; Bonaventura, A.; Vecchiè, A.; Dallegri, F.; Carbone, F. The Role of Inflammation in Cardiovascular Outcome. Curr. Atheroscler. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. JCM 2019, 8, 2072. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 Inflammasome as a Key Driver of Vascular Disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-Cell Immune Landscape of Human Atherosclerotic Plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An Inflammatory Disease. New Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Kritharides, L. Inflammatory Markers and Outcomes in Cardiovascular Disease. PLoS Med. 2009, 6, e1000147. [Google Scholar] [CrossRef] [PubMed]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Fu, Y.; Yuan, J.; Sang, F.; Shao, M.; Yan, S.; Li, L.; Zhu, R.; Wang, Z. Biejiajian Pill Ameliorates Diabetes-Associated Atherosclerosis through Inhibition of the NLRP3 Inflammasome. Evid. -Based Complement. Altern. Med. 2022, 2022, 9131178. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 Inflammasome Promotes Diabetes-Induced Endothelial Inflammation and Atherosclerosis. DMSO 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. IJMS 2019, 20, 3328. [Google Scholar] [CrossRef]
- Fatima, S.; Ambreen, S.; Mathew, A.; Elwakiel, A.; Gupta, A.; Singh, K.; Krishnan, S.; Rana, R.; Khawaja, H.; Gupta, D.; et al. ER-Stress and Senescence Coordinately Promote Endothelial Barrier Dysfunction in Diabetes-Induced Atherosclerosis. Nutrients 2022, 14, 2786. [Google Scholar] [CrossRef]
- Mikkelsen, R.R.; Hundahl, M.P.; Torp, C.K.; Rodríguez-Carrio, J.; Kjolby, M.; Bruun, J.M.; Kragstrup, T.W. Immunomodulatory and Immunosuppressive Therapies in Cardiovascular Disease and Type 2 Diabetes Mellitus: A Bedside-to-Bench Approach. Eur. J. Pharmacol. 2022, 925, 174998. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 10. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. NLRP3: A Novel Mediator in Cardiovascular Disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.-X.; Monaco, C. Therapeutic Strategies Targeting Inflammation and Immunity in Atherosclerosis: How to Proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef]
- Lee, J.; Wan, J.; Lee, L.; Peng, C.; Xie, H.; Lee, C. Study of the NLRP3 Inflammasome Component Genes and Downstream Cytokines in Patients with Type 2 Diabetes Mellitus with Carotid Atherosclerosis. Lipids Health Dis. 2017, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Parsamanesh, N.; Moossavi, M.; Bahrami, A.; Fereidouni, M.; Barreto, G.; Sahebkar, A. NLRP3 Inflammasome as a Treatment Target in Atherosclerosis: A Focus on Statin Therapy. Int. Immunopharmacol. 2019, 73, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 Inflammasome Pathways in Atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef]
- Tong, Y.; Wang, Z.; Cai, L.; Lin, L.; Liu, J.; Cheng, J. NLRP3 Inflammasome and Its Central Role in the Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4293206. [Google Scholar] [CrossRef]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Trans. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid. -Based Complement. Altern. Med. 2020, 2020, 1561342. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Yu, Y.; Li, M.; Chen, R. NLRP3 Inflammasome, an Immune-inflammatory Target in Pathogenesis and Treatment of Cardiovascular Diseases. Clin. Transl. Med. 2020, 10, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, D.; Huo, X.; Wu, Y.; Liu, C.; Sun, Q. Pyroptosis in NLRP3 Inflammasome-Related Atherosclerosis. CST 2022, 6, 79–88. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and Atherosclerosis: Signaling Pathways and Therapeutic Intervention. Sig. Transduct. Target 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- González-Moro, A.; Valencia, I.; Shamoon, L.; Sánchez-Ferrer, C.F.; Peiró, C.; de la Cuesta, F. NLRP3 Inflammasome in Vascular Disease: A Recurrent Villain to Combat Pharmacologically. Antioxidants 2022, 11, 269. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 Inflammasome Activation and Cell Death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Moasses Ghafary, S.; Soriano-Teruel, P.M.; Lotfollahzadeh, S.; Sancho, M.; Serrano-Candelas, E.; Karami, F.; Barigye, S.J.; Fernández-Pérez, I.; Gozalbes, R.; Nikkhah, M.; et al. Identification of NLRP3PYD Homo-Oligomerization Inhibitors with Anti-Inflammatory Activity. IJMS 2022, 23, 1651. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 Inflammasome: Its Regulation and Involvement in Atherosclerosis. J. Cell Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, L.; Dong, N.; Li, F. NLRP3 Inflammasome: The Rising Star in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 927061. [Google Scholar] [CrossRef]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, S.; Yang, G.; Wang, N. Spotlight on NLRP3 Inflammasome: Role in Pathogenesis and Therapies of Atherosclerosis. JIR 2021, 14, 7143–7172. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.; Yu, T.; Chu, X. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on Dispersed Trans-Golgi Network Mediates NLRP3 Inflammasome Activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural Mechanism for NEK7-Licensed Activation of NLRP3 Inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroder, K. Inflammasome Signaling and Regulation of Interleukin-1 Family Cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. JAHA 2019, 8, e012219. [Google Scholar] [CrossRef]
- Pellegrini, C.; Martelli, A.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Calderone, V. NLRP3 Inflammasome in Cardiovascular Diseases: Pathophysiological and Pharmacological Implications. Med. Res. Rev. 2021, 41, 1890–1926. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-C.; Chang, Y.-H. Comparison of Inhibitory Capacities of 6-, 8- and 10-Gingerols/Shogaols on the Canonical NLRP3 Inflammasome-Mediated IL-1β Secretion. Molecules 2018, 23, 466. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Mu, Q.; Bao, X.; Zuo, J.; Fang, X.; Hua, J.; Zhang, D.; Jiang, G.; Li, P.; Gao, S.; et al. Targeting NLRP3 Inflammasome in the Treatment Of Diabetes and Diabetic Complications: Role of Natural Compounds from Herbal Medicine. Aging Dis. 2021, 12, 1587. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1812. [Google Scholar] [CrossRef]
- Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. IJMS 2021, 22, 3254. [Google Scholar] [CrossRef]
- Hasheminasabgorji, E.; Jha, J.C. Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors. Biomedicines 2021, 9, 1602. [Google Scholar] [CrossRef]
- Pickering, R.J.; Rosado, C.J.; Sharma, A.; Buksh, S.; Tate, M.; de Haan, J.B. Recent Novel Approaches to Limit Oxidative Stress and Inflammation in Diabetic Complications. Clin. Transl. Immunol. 2018, 7, e1016. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. IJMS 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Sterol Regulatory Element Binding Protein (SREBP) -1 Mediates Oxidized Low-Density Lipoprotein (OxLDL) Induced Macrophage Foam Cell Formation through NLRP3 Inflammasome Activation. Cell Signal. 2019, 53, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, S. NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention. IJMS 2021, 22, 13228. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, H.; Yang, Y.; Wang, H. The Role of H2S Regulating NLRP3 Inflammasome in Diabetes. IJMS 2022, 23, 4818. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Xing, Q. NLRP3 Inflammasomes Show High Expression in Aorta of Patients with Atherosclerosis. Heart Lung Circ. 2013, 22, 746–750. [Google Scholar] [CrossRef]
- Shi, X.; Xie, W.-L.; Kong, W.-W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. JAHA 2016, 5, e003031. [Google Scholar] [CrossRef]
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with Severity and Prognosis of Coronary Atherosclerosis in Acute Coronary Syndrome Patients. Heart Vessel. 2016, 31, 1218–1229. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 Inflammasomes Contributes to Hyperhomocysteinemia-Aggravated Inflammation and Atherosclerosis in ApoE-Deficient Mice. Lab. Investig. 2017, 97, 922–934. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef]
- Abderrazak, A.; Couchie, D.; Darweesh Mahmood, D.F.; Elhage, R.; Vindis, C.; Laffargue, M.; Matéo, V.; Büchele, B.; Ayala, M.R.; Gaafary, M.E.; et al. Response to Letter Regarding Article, “Anti-Inflammatory and Antiatherogenic Effects of the Inflammasome NLRP3 Inhibitor Arglabin in ApoE2. Ki Mice Fed a High-Fat Diet.” Circulation 2015, 132, e250–e251. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxidative Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report. ATVB 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Yang, Y.; Ou, T.; Key, C.-C.C.; Tong, S.H.; Sequeira, R.C.; Nelson, J.M.; Nie, Y.; Wang, Z.; Boudyguina, E.; et al. Dietary PUFAs Attenuate NLRP3 Inflammasome Activation via Enhancing Macrophage Autophagy. J. Lipid Res. 2017, 58, 1808–1821. [Google Scholar] [CrossRef]
- Yin, R.; Zhu, X.; Wang, J.; Yang, S.; Ma, A.; Xiao, Q.; Song, J.; Pan, X. MicroRNA-155 Promotes the Ox-LDL-Induced Activation of NLRP3 Inflammasomes via the ERK1/2 Pathway in THP-1 Macrophages and Aggravates Atherosclerosis in ApoE−/− Mice. Ann. Palliat. Med. 2019, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 Deficiency Decreases Atherosclerosis in Apolipoprotein E-Null Mice. Can. J. Cardiol. 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine Promotes Atherosclerosis via ROS-NLRP3-Mediated Endothelial Cell Pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Bai, X.; Lin, Y.; Li, Z.; Fu, J.; Li, M.; Zhao, T.; Yang, H.; Xu, R.; et al. Melatonin Prevents Endothelial Cell Pyroptosis via Regulation of Long Noncoding RNA MEG3/MiR-223/NLRP3 Axis. J. Pineal Res. 2018, 64, e12449. [Google Scholar] [CrossRef]
- Li, P.; Zhong, X.; Li, J.; Liu, H.; Ma, X.; He, R.; Zhao, Y. MicroRNA-30c-5p Inhibits NLRP3 Inflammasome-Mediated Endothelial Cell Pyroptosis through FOXO3 down-Regulation in Atherosclerosis. Biochem. Biophys. Res. Commun. 2018, 503, 2833–2840. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.S.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, S.; Kotlyarova, A. Molecular Pharmacology of Inflammation Resolution in Atherosclerosis. IJMS 2022, 23, 4808. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharm. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. IJMS 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2021, 10, 18. [Google Scholar] [CrossRef]
- Haas, A.V.; McDonnell, M.E. Pathogenesis of Cardiovascular Disease in Diabetes. Endocrinol. Metab. Clin. North. Am. 2018, 47, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants 2022, 11, 1958. [Google Scholar] [CrossRef]
- Li, J.; Fu, X.; Yang, R.; Zhang, W. Atherosclerosis Vascular Endothelial Secretion Dysfunction and Smooth Muscle Cell Proliferation. J. Healthc. Eng. 2022, 2022, 9271879. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial Responses to Shear Stress in Atherosclerosis: A Novel Role for Developmental Genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. (Landmark Ed) 2022, 27, 0105. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Janjusevic, M.; Fluca, A.L.; Gagno, G.; Pierri, A.; Padoan, L.; Sorrentino, A.; Beltrami, A.P.; Sinagra, G.; Aleksova, A. Old and Novel Therapeutic Approaches in the Management of Hyperglycemia, an Important Risk Factor for Atherosclerosis. IJMS 2022, 23, 2336. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.-K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef]
- Beverly, J.K.; Budoff, M.J. Atherosclerosis: Pathophysiology of Insulin Resistance, Hyperglycemia, Hyperlipidemia, and Inflammation. J. Diabetes 2020, 12, 102–104. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Lei, Y.; Tzvetkov, N.T.; Liu, X.; Yeung, A.W.K.; Xu, S.; Atanasov, A.G. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharm. Rev. 2019, 71, 596–670. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, M.; Zeng, L.; Wang, D. The Beneficial Role of Nrf2 in the Endothelial Dysfunction of Atherosclerosis. Cardiol. Res. Pract. 2022, 2022, 4287711. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wang, C.; Jin, Y.; Meng, Q.; Wu, J.; Sun, H. Kaempferol-Induced GPER Upregulation Attenuates Atherosclerosis via the PI3K/AKT/Nrf2 Pathway. Pharm. Biol. 2021, 59, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Samarghandian, S.; Azimi-Nezhad, M.; Farkhondeh, T. Immunomodulatory and Antioxidant Effects of Saffron Aqueous Extract (Crocus Sativus L.) on Streptozotocin-Induced Diabetes in Rats. Indian Heart J. 2017, 69, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Lecis, D. Inflammation in Atherosclerotic Cardiovascular Disease. F1000Res 2019, 8, 1402. [Google Scholar] [CrossRef]
- Drareni, K.; Gautier, J.-F.; Venteclef, N.; Alzaid, F. Transcriptional Control of Macrophage Polarisation in Type 2 Diabetes. Semin. Immunopathol. 2019, 41, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Ji, H.-H.; Li, Y.-J.; Guo, S.-D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef] [PubMed]
- Niyonzima, N.; Bakke, S.S.; Gregersen, I.; Holm, S.; Sandanger, Ø.; Orrem, H.L.; Sporsheim, B.; Ryan, L.; Kong, X.Y.; Dahl, T.B.; et al. Cholesterol Crystals Use Complement to Increase NLRP3 Signaling Pathways in Coronary and Carotid Atherosclerosis. EBioMedicine 2020, 60, 102985. [Google Scholar] [CrossRef]
- Qi, H.-M.; Cao, Q.; Liu, Q. TLR4 Regulates Vascular Smooth Muscle Cell Proliferation in Hypertension. Am. J. Transl. Res. 2021, 13, 314–325. [Google Scholar]
- Poznyak, A.V.; Melnichenko, A.A.; Wetzker, R.; Gerasimova, E.V.; Orekhov, A.N. NLPR3 Inflammasomes and Their Significance for Atherosclerosis. Biomedicines 2020, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Catapano, A.L.; Magni, P. Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease. IJMS 2020, 21, 4459. [Google Scholar] [CrossRef]
- Hill, J.R.; Coll, R.C.; Sue, N.; Reid, J.C.; Dou, J.; Holley, C.L.; Pelingon, R.; Dickinson, J.B.; Biden, T.J.; Schroder, K.; et al. Sulfonylureas as Concomitant Insulin Secretagogues and NLRP3 Inflammasome Inhibitors. ChemMedChem 2017, 12, 1449–1457. [Google Scholar] [CrossRef]
- Ngo, L.; Oh, J.; Kim, A.; Back, H.; Kang, W.; Chae, J.; Yun, H.; Lee, H. Development of a Pharmacokinetic Model Describing Neonatal Fc Receptor-Mediated Recycling of HL2351, a Novel Hybrid Fc-Fused Interleukin-1 Receptor Antagonist, to Optimize Dosage Regimen. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 584–595. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/MTOR-Dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Liu, Y.; Rundberg Nilsson, A.; Gatchalian, R.; Crother, T.R.; Tourtellotte, W.G.; Zhang, Y.; Aleman-Muench, G.R.; Lewis, G.; Chen, W.; et al. Metformin Inhibition of Mitochondrial ATP and DNA Synthesis Abrogates NLRP3 Inflammasome Activation and Pulmonary Inflammation. Immunity 2021, 54, 1463–1477.e11. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, Macrophage Dysfunction and Atherosclerosis. Front. Immunol. 2021, 12, 682853. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, L.; Zhong, X.; Yue, Y.; Hong, Y.; Li, Y.; Li, Y. Metformin Reduced NLRP3 Inflammasome Activity in Ox-LDL Stimulated Macrophages through Adenosine Monophosphate Activated Protein Kinase and Protein Phosphatase 2A. Eur. J. Pharmacol. 2019, 852, 99–106. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin Inhibited Nod-like Receptor Protein 3 Inflammasomes Activation and Suppressed Diabetes-Accelerated Atherosclerosis in ApoE−/− Mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 Inhibition Modulates NLRP3 Inflammasome Activity via Ketones and Insulin in Diabetes with Cardiovascular Disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Chen, H.; Tran, D.; Yang, H.-C.; Nylander, S.; Birnbaum, Y.; Ye, Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: An AMPK–MTOR Interplay. Cardiovasc. Drugs 2020, 34, 443–461. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of Sodium Glucose Cotransporter 2 (SGLT2) Inhibitors on Atherosclerosis: From Pharmacology to Pre-Clinical and Clinical Therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef] [PubMed]
- Razavi, M.; Wei, Y.-Y.; Rao, X.-Q.; Zhong, J.-X. DPP-4 Inhibitors and GLP-1RAs: Cardiovascular Safety and Benefits. Mil. Med. Res. 2022, 9, 45. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs 2017, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Du, X.; Yao, X.; Zhao, Y. Vildagliptin Inhibits High Free Fatty Acid (FFA)-Induced NLRP3 Inflammasome Activation in Endothelial Cells. Artif. Cell. Nanomed. Biotechnol. 2019, 47, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Jiang, D.; Zhang, L.; Ding, M.; Zhou, H. Anagliptin Ameliorates High Glucose- Induced Endothelial Dysfunction via Suppression of NLRP3 Inflammasome Activation Mediated by SIRT1. Mol. Immunol. 2019, 107, 54–60. [Google Scholar] [CrossRef]
- Luo, X.; Hu, Y.; He, S.; Ye, Q.; Lv, Z.; Liu, J.; Chen, X. Dulaglutide Inhibits High Glucose- Induced Endothelial Dysfunction and NLRP3 Inflammasome Activation. Arch. Biochem. Biophys. 2019, 671, 203–209. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Corcoran, S.E.; Halai, R.; Cooper, M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharm. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef]
- Mekni, N.; De Rosa, M.; Cipollina, C.; Gulotta, M.R.; De Simone, G.; Lombino, J.; Padova, A.; Perricone, U. In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots. IJMS 2019, 20, 4974. [Google Scholar] [CrossRef]
- Sharma, A.; Choi, J.S.Y.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E.; et al. Specific NLRP3 Inhibition Protects Against Diabetes-Associated Atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Fan, Y.; Xue, G.; Chen, Q.; Lu, Y.; Dong, R.; Yuan, H. CY-09 Inhibits NLRP3 Inflammasome Activation to Relieve Pain via TRPA1. Comput. Math. Methods Med. 2021, 2021, 9806690. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hong, W.; Lu, S.; Li, Y.; Guan, Y.; Weng, X.; Feng, Z. The NLRP3 Inflammasome in Non-Alcoholic Fatty Liver Disease and Steatohepatitis: Therapeutic Targets and Treatment. Front. Pharmacol. 2022, 13, 780496. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 Inflammasome Inhibitor OLT1177 Rescues Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-Sulfonyl Nitrile Compound, Safe in Humans, Inhibits the NLRP3 Inflammasome and Reverses the Metabolic Cost of Inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast Directly Targets NLRP 3 to Treat Inflammasome-driven Diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Cummins, C.B.; Wang, X.; Sommerhalder, C.; Bohanon, F.J.; Nunez Lopez, O.; Tie, H.-Y.; Rontoyanni, V.G.; Zhou, J.; Radhakrishnan, R.S. Natural Compound Oridonin Inhibits Endotoxin-Induced Inflammatory Response of Activated Hepatic Stellate Cells. BioMed Res. Int. 2018, 2018, 6137420. [Google Scholar] [CrossRef]
- Xu, J.; Wold, E.; Ding, Y.; Shen, Q.; Zhou, J. Therapeutic Potential of Oridonin and Its Analogs: From Anticancer and Antiinflammation to Neuroprotection. Molecules 2018, 23, 474. [Google Scholar] [CrossRef]
- Xu, M.; Wan, C.; Huang, S.; Wang, H.; Fan, D.; Wu, H.-M.; Wu, Q.; Ma, Z.; Deng, W.; Tang, Q.-Z. Oridonin Protects against Cardiac Hypertrophy by Promoting P21-Related Autophagy. Cell Death Dis. 2019, 10, 403. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin Is a Covalent NLRP3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, C.-T.; Ma, W.; Xie, X.; Huang, Q. Oridonin: A Review of Its Pharmacology, Pharmacokinetics and Toxicity. Front. Pharmacol. 2021, 12, 645824. [Google Scholar] [CrossRef]
- Zheng, J.; Jiang, Z.; Song, Y.; Huang, S.; Du, Y.; Yang, X.; Xiao, Y.; Ma, Z.; Xu, D.; Li, J. 3,4-Methylenedioxy-β-Nitrostyrene Alleviates Dextran Sulfate Sodium–Induced Mouse Colitis by Inhibiting the NLRP3 Inflammasome. Front. Pharmacol. 2022, 13, 866228. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.; Pellegrini, C.; Martínez-Banaclocha, H.; Giorgis, M.; Marini, E.; Costale, A.; Miglio, G.; Fornai, M.; Antonioli, L.; López-Castejón, G.; et al. Development of an Acrylate Derivative Targeting the NLRP3 Inflammasome for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 3656–3671. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxidative Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef]
- Shi, Y.; Lv, Q.; Zheng, M.; Sun, H.; Shi, F. NLRP3 Inflammasome Inhibitor INF39 Attenuated NLRP3 Assembly in Macrophages. Int. Immunopharmacol. 2021, 92, 107358. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Zhao, J.; Xia, S.; Ruan, J.; Luo, X.; Kim, J.; Lieberman, J.; Wu, H. Identification of Pyroptosis Inhibitors That Target a Reactive Cysteine in Gasdermin D. Nat. Immunol. 2018, 7, A132. [Google Scholar]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Li, Y.; Niu, X.; Xu, H.; Li, Q.; Meng, L.; He, M.; Zhang, J.; Zhang, Z.; Zhang, Z. VX-765 Attenuates Atherosclerosis in ApoE Deficient Mice by Modulating VSMCs Pyroptosis. Exp. Cell Res. 2020, 389, 111847. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.L.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 Blockade With Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; Bax, W.A.; Budgeon, C.A.; Tijssen, J.G.P.; Mosterd, A.; Cornel, J.H.; et al. The Effect of Low-Dose Colchicine in Patients with Stable Coronary Artery Disease: The LoDoCo2 Trial Rationale, Design, and Baseline Characteristics. Am. Heart J. 2019, 218, 46–56. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef]
- Lee, H.G.; Cho, N.; Jeong, A.J.; Li, Y.-C.; Rhie, S.-J.; Choi, J.S.; Lee, K.-H.; Kim, Y.; Kim, Y.-N.; Kim, M.-H.; et al. Immunomodulatory Activities of the Benzoxathiole Derivative BOT-4-One Ameliorate Pathogenic Skin Inflammation in Mice. J. Investig. Dermatol. 2016, 136, 107–116. [Google Scholar] [CrossRef]
- Ahn, H.; Kang, S.G.; Yoon, S.; Ko, H.-J.; Kim, P.-H.; Hong, E.-J.; An, B.-S.; Lee, E.; Lee, G.-S. Methylene Blue Inhibits NLRP3, NLRC4, AIM2, and Non-Canonical Inflammasome Activation. Sci. Rep. 2017, 7, 12409. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, H.; Yu, J.; Chen, X.; Yang, L. Methylene Blue Attenuates Diabetic Retinopathy by Inhibiting NLRP3 Inflammasome Activation in STZ-Induced Diabetic Rats. Ocul. Immunol. Inflamm. 2019, 27, 836–843. [Google Scholar] [CrossRef]
- Deng, W.; Yang, Z.; Yue, H.; Ou, Y.; Hu, W.; Sun, P. Disulfiram Suppresses NLRP3 Inflammasome Activation to Treat Peritoneal and Gouty Inflammation. Free Radic. Biol. Med. 2020, 152, 8–17. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs Inhibit the NLRP3 Inflammasome and Protect against Alzheimer’s Disease in Rodent Models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Ambati, M.; Apicella, I.; Wang, S.; Narendran, S.; Leung, H.; Pereira, F.; Nagasaka, Y.; Huang, P.; Varshney, A.; Baker, K.L.; et al. Identification of Fluoxetine as a Direct NLRP3 Inhibitor to Treat Atrophic Macular Degeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2102975118. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, T.M.; Inzaugarat, M.E.; Knorr, J.; Geisler, L.; Schulz, M.; Bieghs, V.; Frissen, M.; Feldstein, A.E.; Tacke, F.; Trautwein, C.; et al. Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner. Cells 2021, 10, 2618. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The Angiotensin-(1-7)/Mas Receptor Axis Protects from Endothelial Cell Senescence via Klotho and Nrf2 Activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- McKie, E.A.; Reid, J.L.; Mistry, P.C.; DeWall, S.L.; Abberley, L.; Ambery, P.D.; Gil-Extremera, B. A Study to Investigate the Efficacy and Safety of an Anti-Interleukin-18 Monoclonal Antibody in the Treatment of Type 2 Diabetes Mellitus. PLoS ONE 2016, 11, e0150018. [Google Scholar] [CrossRef]
- Klein, A.L.; Imazio, M.; Cremer, P.; Brucato, A.; Abbate, A.; Fang, F.; Insalaco, A.; LeWinter, M.; Lewis, B.S.; Lin, D.; et al. Phase 3 Trial of Interleukin-1 Trap Rilonacept in Recurrent Pericarditis. N. Engl. J. Med. 2021, 384, 31–41. [Google Scholar] [CrossRef]
- Wu, L.-M.; Wu, S.-G.; Chen, F.; Wu, Q.; Wu, C.-M.; Kang, C.-M.; He, X.; Zhang, R.-Y.; Lu, Z.-F.; Li, X.-H.; et al. Atorvastatin Inhibits Pyroptosis through the LncRNA NEXN-AS1/NEXN Pathway in Human Vascular Endothelial Cells. Atherosclerosis 2020, 293, 26–34. [Google Scholar] [CrossRef]
- Peng, S.; Xu, L.-W.; Che, X.-Y.; Xiao, Q.-Q.; Pu, J.; Shao, Q.; He, B. Atorvastatin Inhibits Inflammatory Response, Attenuates Lipid Deposition, and Improves the Stability of Vulnerable Atherosclerotic Plaques by Modulating Autophagy. Front. Pharmacol. 2018, 9, 438. [Google Scholar] [CrossRef]
- Kong, F.; Ye, B.; Lin, L.; Cai, X.; Huang, W.; Huang, Z. Atorvastatin Suppresses NLRP3 Inflammasome Activation via TLR4/MyD88/NF-ΚB Signaling in PMA-Stimulated THP-1 Monocytes. Biomed. Pharmacother. 2016, 82, 167–172. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, S.; Hu, S.; Li, H.; Li, M.; Geng, X.; Wang, H. NLRP3 Inflammasome Expression in Peripheral Blood Monocytes of Coronary Heart Disease Patients and Its Modulation by Rosuvastatin. Mol. Med. Rep. 2019, 20, 1826–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jin, L.; Zhuang, Y.; Hu, Y.; Cang, J.; Guo, K. Cardioprotective Effect of Rosuvastatin against Isoproterenol-Induced Myocardial Infarction Injury in Rats. Int. J. Mol. Med. 2018, 41, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Min, D.S.; Park, H.; Kim, H.P. Flavonoids Interfere with NLRP3 Inflammasome Activation. Toxicol. Appl. Pharmacol. 2018, 355, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Hashiguchi, K.; Yamamoto, H.; Tagami, M. Dietary Apigenin Reduces Induction of LOX-1 and NLRP3 Expression, Leukocyte Adhesion, and Acetylated Low-Density Lipoprotein Uptake in Human Endothelial Cells Exposed to Trimethylamine-N-Oxide. J. Cardiovasc. Pharmacol. 2019, 74, 558–565. [Google Scholar] [CrossRef]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. IJMS 2019, 20, 1305. [Google Scholar] [CrossRef]
- Wang, K.; Lv, Q.; Miao, Y.; Qiao, S.; Dai, Y.; Wei, Z. Cardamonin, a Natural Flavone, Alleviates Inflammatory Bowel Disease by the Inhibition of NLRP3 Inflammasome Activation via an AhR/Nrf2/NQO1 Pathway. Biochem. Pharmacol. 2018, 155, 494–509. [Google Scholar] [CrossRef]
- Liao, N.-C.; Shih, Y.-L.; Chou, J.-S.; Chen, K.-W.; Chen, Y.-L.; Lee, M.-H.; Peng, S.-F.; Leu, S.-J.; Chung, J.-G. Cardamonin Induces Cell Cycle Arrest, Apoptosis and Alters Apoptosis Associated Gene Expression in WEHI-3 Mouse Leukemia Cells. Am. J. Chin. Med. 2019, 47, 635–656. [Google Scholar] [CrossRef]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin Is a Potent Inhibitor of NLRP3 Inflammasome Activation and Diet-induced Adipose Tissue Inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M.; et al. Luteolin, a Flavonoid, as an Anticancer Agent: A Review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-C.; Li, Z.; Xu, W.; Xiang, C.-H.; Ma, Y.-F. Luteolin Alleviates NLRP3 Inflammasome Activation and Directs Macrophage Polarization in Lipopolysaccharide-Stimulated RAW264.7 Cells. Am. J. Transl. Res. 2018, 10, 265–273. [Google Scholar] [PubMed]
- Li, B.; Du, P.; Du, Y.; Zhao, D.; Cai, Y.; Yang, Q.; Guo, Z. Luteolin Alleviates Inflammation and Modulates Gut Microbiota in Ulcerative Colitis Rats. Life Sci. 2021, 269, 119008. [Google Scholar] [CrossRef] [PubMed]
- Domiciano, T.P.; Wakita, D.; Jones, H.D.; Crother, T.R.; Verri, W.A.; Arditi, M.; Shimada, K. Quercetin Inhibits Inflammasome Activation by Interfering with ASC Oligomerization and Prevents Interleukin-1 Mediated Mouse Vasculitis. Sci. Rep. 2017, 7, 41539. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Huang, Y.; Han, N.; He, F.; Li, M.; Bian, Z.; Liu, J.; Sun, T.; Zhu, L. Quercetin Suppresses NLRP3 Inflammasome Activation and Attenuates Histopathology in a Rat Model of Spinal Cord Injury. Spinal Cord 2016, 54, 592–596. [Google Scholar] [CrossRef]
- Chen, P.; Bai, Q.; Wu, Y.; Zeng, Q.; Song, X.; Guo, Y.; Zhou, P.; Wang, Y.; Liao, X.; Wang, Q.; et al. The Essential Oil of Artemisia Argyi H.Lév. and Vaniot Attenuates NLRP3 Inflammasome Activation in THP-1 Cells. Front. Pharmacol. 2021, 12, 712907. [Google Scholar] [CrossRef]
- Kwak, S.-B.; Koppula, S.; In, E.-J.; Sun, X.; Kim, Y.-K.; Kim, M.-K.; Lee, K.-H.; Kang, T.-B. Artemisia Extract Suppresses NLRP3 and AIM2 Inflammasome Activation by Inhibition of ASC Phosphorylation. Mediat. Inflamm. 2018, 2018, 6054069. [Google Scholar] [CrossRef]
- Jiang, Y.; Du, H.; Liu, X.; Fu, X.; Li, X.; Cao, Q. Artemisinin Alleviates Atherosclerotic Lesion by Reducing Macrophage Inflammation via Regulation of AMPK/NF-ΚB/NLRP3 Inflammasomes Pathway. J. Drug Target. 2020, 28, 70–79. [Google Scholar] [CrossRef]
- Lee, H.E.; Yang, G.; Kim, N.D.; Jeong, S.; Jung, Y.; Choi, J.Y.; Park, H.H.; Lee, J.Y. Targeting ASC in NLRP3 Inflammasome by Caffeic Acid Phenethyl Ester: A Novel Strategy to Treat Acute Gout. Sci. Rep. 2016, 6, 38622. [Google Scholar] [CrossRef]
- Kong, F.; Ye, B.; Cao, J.; Cai, X.; Lin, L.; Huang, S.; Huang, W.; Huang, Z. Curcumin Represses NLRP3 Inflammasome Activation via TLR4/MyD88/NF-ΚB and P2X7R Signaling in PMA-Induced Macrophages. Front. Pharmacol. 2016, 7, 369. [Google Scholar] [CrossRef]
- Rahban, M.; Habibi-Rezaei, M.; Mazaheri, M.; Saso, L.; Moosavi-Movahedi, A.A. Anti-Viral Potential and Modulation of Nrf2 by Curcumin: Pharmacological Implications. Antioxidants 2020, 9, 1228. [Google Scholar] [CrossRef]
- Gao, J.; Peng, S.; Shan, X.; Deng, G.; Shen, L.; Sun, J.; Jiang, C.; Yang, X.; Chang, Z.; Sun, X.; et al. Inhibition of AIM2 Inflammasome-Mediated Pyroptosis by Andrographolide Contributes to Amelioration of Radiation-Induced Lung Inflammation and Fibrosis. Cell Death Dis. 2019, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.E.; Le, T.H. Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients 2021, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yao, X.; Zhang, Y.; Tang, J. Synthesis, Biological Function and Evaluation of Shikonin in Cancer Therapy. Fitoterapia 2019, 134, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Yang, Q.; Chen, J.; Yu, C.; Zhang, L.; Zhou, W.; Chen, M. Salvianolic Acid A Ameliorates Early-Stage Atherosclerosis Development by Inhibiting NLRP3 Inflammasome Activation in Zucker Diabetic Fatty Rats. Molecules 2020, 25, 1089. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-C.; Yen, C.-K.; Lu, Y.-C.; Shi, C.-S.; Hsieh, R.-Z.; Chang, S.-F.; Chen, C.-N. The Antagonism of 6-Shogaol in High-Glucose-Activated NLRP3 Inflammasome and Consequent Calcification of Human Artery Smooth Muscle Cells. Cell Biosci. 2020, 10, 5. [Google Scholar] [CrossRef]
- Lian, D.; Yuan, H.; Yin, X.; Wu, Y.; He, R.; Huang, Y.; Chen, Y. Puerarin Inhibits Hyperglycemia-Induced Inter-Endothelial Junction through Suppressing Endothelial Nlrp3 Inflammasome Activation via ROS-Dependent Oxidative Pathway. Phytomedicine 2019, 55, 310–319. [Google Scholar] [CrossRef]
- Hu, R.; Wang, M.; Ni, S.; Wang, M.; Liu, L.; You, H.; Wu, X.; Wang, Y.; Lu, L.; Wei, L. Salidroside Ameliorates Endothelial Inflammation and Oxidative Stress by Regulating the AMPK/NF-ΚB/NLRP3 Signaling Pathway in AGEs-Induced HUVECs. Eur. J. Pharmacol. 2020, 867, 172797. [Google Scholar] [CrossRef]
- Feng, H.; Zhu, X.; Tang, Y.; Fu, S.; Kong, B.; Liu, X. Astragaloside IV Ameliorates Diabetic Nephropathy in Db/Db Mice by Inhibiting NLRP3 Inflammasome-mediated Inflammation. Int. J. Mol. Med. 2021, 48, 164. [Google Scholar] [CrossRef]
- Qian, W.; Cai, X.; Qian, Q.; Zhuang, Q.; Yang, W.; Zhang, X.; Zhao, L. Astragaloside IV Protects Endothelial Progenitor Cells from the Damage of Ox-LDL via the LOX-1/NLRP3 Inflammasome Pathway. DDDT 2019, 13, 2579–2589. [Google Scholar] [CrossRef]
- Burger, F.; Baptista, D.; Roth, A.; Da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. IJMS 2021, 23, 340. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The Selective NLRP3 Inhibitor MCC950 Hinders Atherosclerosis Development by Attenuating Inflammation and Pyroptosis in Macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef] [PubMed]


| Agent | Study Design | Salient Results | Ref. |
|---|---|---|---|
| Phase I (priming) | |||
| Bay 11-7082 | Preclinical (Experimental study) | - Via NF-κB inhibition selectively blocks IKKβ kinase activity with subsequent inhibition of the NLRP3 inflammasome activation; - Inhibited nigericin-induced and MSU-induced caspase-1 activation by the NLRP3 inflammasome. | [148] |
| NLRP3 oligomerization | |||
| CY-09 (glitazone derivate) | Preclinical (Experimental study) | - It binds to the ATP-binding motif of NLRP3 NACHT domain and inhibits NLRP3 ATPase assembly and activity in macrophages; - Inhibits NLRP3 ATPase activity; - Reverses metabolic effects via NLRP3 inhibition. | [166] |
| MCC 950 | Preclinical (Experimental study) | - Inhibits NLRP3 inflammasome activation by suppressing IL-1β secretion; - It does not affect NLRP1, NLRC4 or AIM2 inflammasomes; - Atheroprotective activity by reducing the size of the plaque; - Reversed the impaired endothelial dysfunction. | [129,130,131] |
| Dapansutrile | CT (Phase I, randomized controlled trial) | - Inhibits NLRP3-ASC band NLRP3-caspase-1 interaction. | [137] |
| Tranilast | Approved (Experimental study) | - Reduces ROS, TXNIP expression and directly inhibits xhantine oxidase activity in vitro; - Via binding to the NATCH domain of NLRP3, inhibits assembly and its effects. | [21,36] |
| Phase II (activation) | |||
| Ang-(1-7) | Preclinical (Experimental study) | - Anti-inflammatory and anti-senescent action through RAAS; - Inhibits IL-1 -induced iNOS expression and NF-κB activation in vascular smooth muscle cells; - Diminishes NLRP3 inflammasome/IL-1 over-activation loop. | [167] |
| HL2351 | CT (Phase I, randomized controlled trial) | -Inhibition of IL-1 function with indirect NLRP3 inflammasome action. | [113] |
| GSK1070806 | CT (Phase II, randomized, placebo-controlled) | - Inhibition of IL-18; - Inhibition of IL-18 did not lead to any improvements in glucose control. | [168] |
| Rilonacept | Approved (Phase III, double-blind, randomized-withdrawal) | - Inhibition of the IL-1 pathway; - Reduced the activation of endothelial cell NADPH oxidase. | [169] |
| Canakinumab | Approved (Randomized, double-blind) | - Direct blockade of IL-1 or its receptor; - Antioxidant effects. | [11] |
| Anakinra | Approved (Randomized, double-blind) | - Modulation of mitochondrial ROS production by activating SOD2. | [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanase, D.M.; Valasciuc, E.; Gosav, E.M.; Ouatu, A.; Buliga-Finis, O.N.; Floria, M.; Maranduca, M.A.; Serban, I.L. Portrayal of NLRP3 Inflammasome in Atherosclerosis: Current Knowledge and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 8162. https://doi.org/10.3390/ijms24098162
Tanase DM, Valasciuc E, Gosav EM, Ouatu A, Buliga-Finis ON, Floria M, Maranduca MA, Serban IL. Portrayal of NLRP3 Inflammasome in Atherosclerosis: Current Knowledge and Therapeutic Targets. International Journal of Molecular Sciences. 2023; 24(9):8162. https://doi.org/10.3390/ijms24098162
Chicago/Turabian StyleTanase, Daniela Maria, Emilia Valasciuc, Evelina Maria Gosav, Anca Ouatu, Oana Nicoleta Buliga-Finis, Mariana Floria, Minela Aida Maranduca, and Ionela Lacramioara Serban. 2023. "Portrayal of NLRP3 Inflammasome in Atherosclerosis: Current Knowledge and Therapeutic Targets" International Journal of Molecular Sciences 24, no. 9: 8162. https://doi.org/10.3390/ijms24098162