
Compartmental Cerebrospinal Fluid Events Occurring after Subarachnoid Hemorrhage: An “Heparin Oriented” Systematic Review
 , and
, and 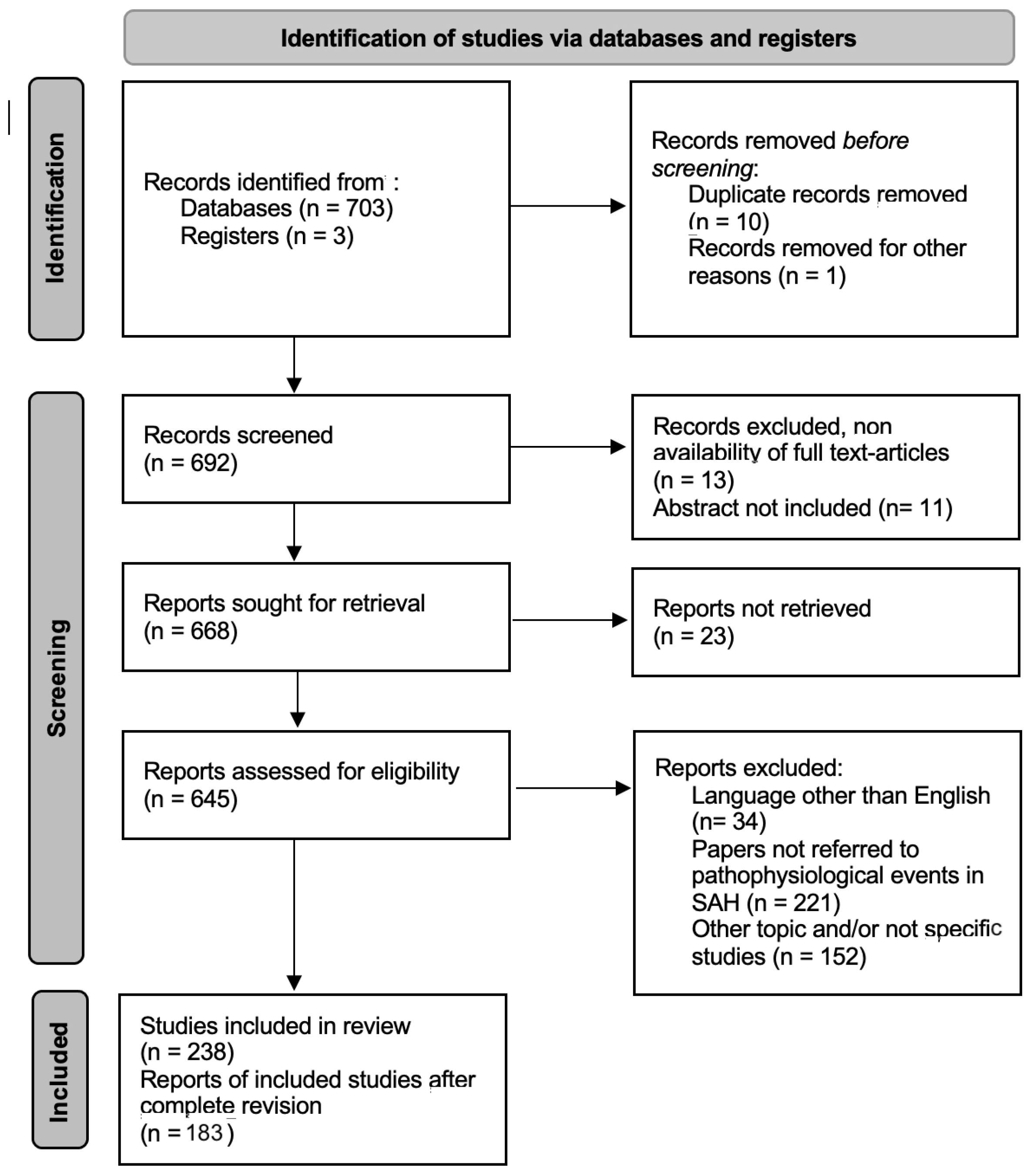{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
- -
- Meta-analysis, Case series, or Clinical study reporting cases of subarachnoid hemorrhage with measurement of CSF inflammation mediators involved in the pathogenesis of DCI.
- (Subarachnoid hemorrhage, CSF) AND (hemoglobin degradation products), 106 articles;
- (Subarachnoid hemorrhage, CSF) AND (platelets), 43 articles;
- (Subarachnoid hemorrhage, CSF) AND (complement), 18 articles;
- (Subarachnoid hemorrhage, CSF) AND (cytokines), 117 articles;
- (Subarachnoid hemorrhage, CSF) AND (chemokines), 15 articles;
- (Subarachnoid hemorrhage, CSF) AND (monocytes), 16 articles;
- (Subarachnoid hemorrhage, CSF) AND (leucocytes), 55 articles;
- (Subarachnoid hemorrhage, CSF) AND (endothelin-1), 39 articles;
- (Subarachnoid hemorrhage, CSF) AND (NO-synthase), 27 articles;
- (Subarachnoid hemorrhage, CSF) AND (ostepontin), 28 articles;
- (Subarachnoid hemorrhage) AND (excitotoxicity), 55 articles
- (Subarachnoid hemorrhage, CSF) AND (matricellular proteins), 36 articles;
- (Subarachnoid hemorrhage, CSF) AND (blood-brain barrier disruption), 61 articles;
- (Subarachnoid hemorrhage, CSF) AND (microglia polarization), 17 articles;
- (Subarachnoid hemorrhage, CSF) AND (heparin), 73 articles.
- -
- Availability of full-text articles
- -
- English text only
- -
- Clinical studies with patients older than 18-year-old with a history of SAH
- -
- Full-text articles in languages other than English
- -
- Studies not referred to pathophysiological events
- -
- Patients younger than 18-year-old
3. Results
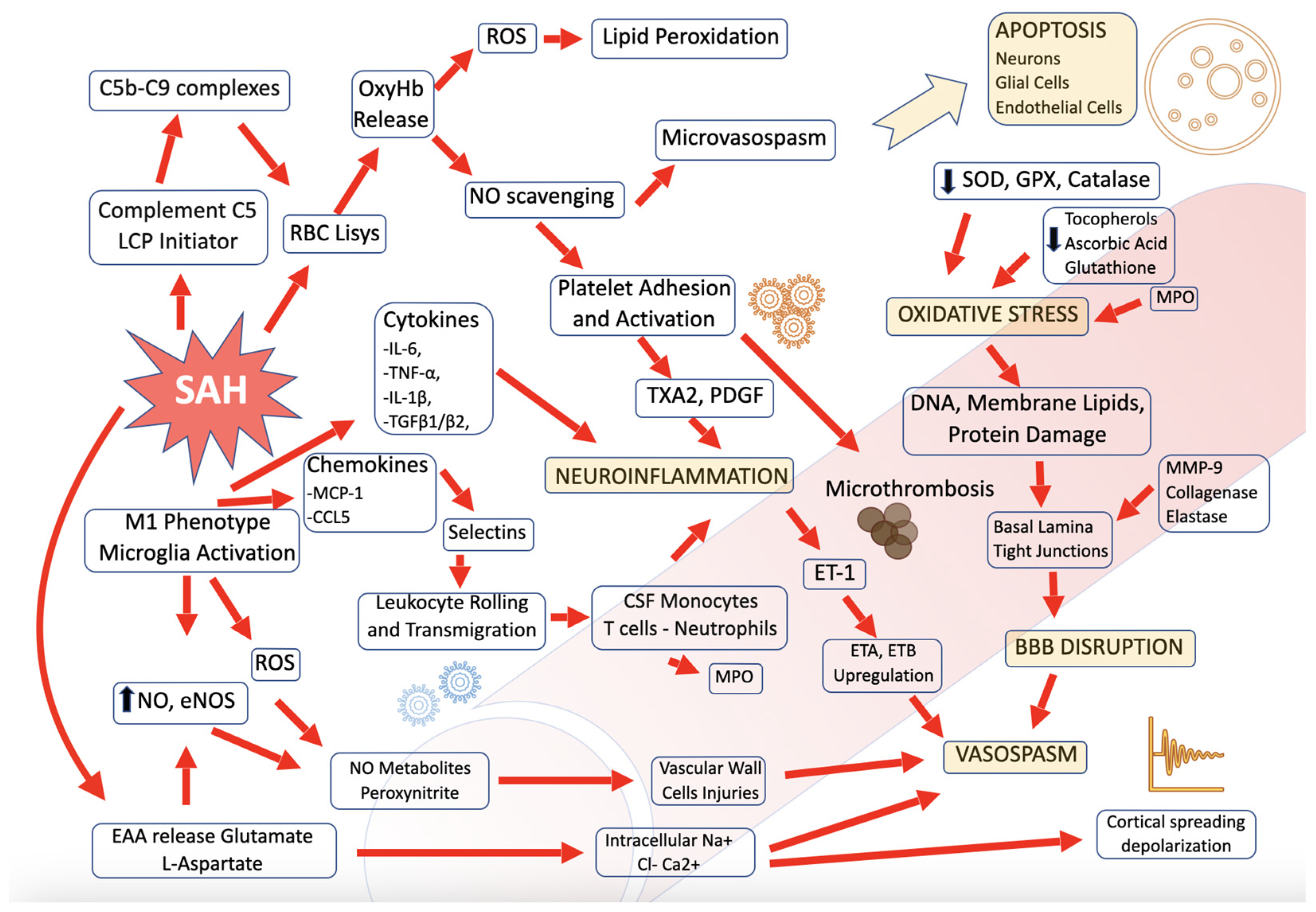
3.1. Hemoglobin Degradation Product and Platelet
3.2. Complement in CSF
3.3. Cytokines in CSF
- -
- -
- CSF IL-6 seems to be a reliable early marker of vasospasm after SAH on day 3 after treatment before vasospasm clinical onset [61].
- -
- CSF IL-6 levels above a cut-off value of 3100 pg/mL are associated with an increased likelihood of ventriculitis; patients with CSF IL-6 levels between 530 and 3100pg/mL are at higher risk for cerebral vasospasm [62].
- -
- An increase of intrathecal IL-6 to values ≥ 10,000 pg/mL in the early post-SAH period may be a useful diagnostic tool to predict shunt dependency after SAH [63].
- -
- IL-6 receptor antagonist Tocilizumab may be evaluated as a therapeutical agent able to significantly reduce microclot formation, neuronal cell death, and delayed cerebral vasospasm [64].
- -
- CSF levels of IL-6 increase over time and are associated with hemorrhage grade. Elevated IL-6 CSF levels may influence SAH progression and may predict poor clinical outcomes in SAH patients. Tumor necrosis factor-α (TNF-α) levels in CSF of SAH patients were higher than those of healthy controls and TNF-α CSF levels increased with disease severity, suggesting that elevated TNF-α levels in CSF may be associated with SAH progression. TNF-α level also correlates with delayed complications of SAH such as DCI [65,66].
- -
- Upregulation of H2S-producing enzymes and IL-6 is associated with the inflammatory response and neurological deficits after SAH [67].
- -
- TGFβ1 and total TGFβ2 increased significantly in adults following SAH, and there was a significant association between higher CSF total TGFβ1/β2 levels in the acute post-hemorrhagic phase and the subsequent development of chronic communicating hydrocephalus [68].
- -
- CSF concentration of Histidine-rich Glycoprotein (HRG) has the possibility to become an early predictor of cerebral vasospasm [69].
- -
- Endothelin-1, IL-6, TNF-α, TNFR-I, and IL-1 receptor antagonist (IL-1ra) is elevated in patients with vasospasm [70].
- -
- CSF levels of IL-6, TNF-α, IL-17A, IL-10, IL-2, and IFN-γ in the early and delayed phase of aSAH patients were increased as compared to controls. IFN-γ and IL-4 were also increased but did not reach statistical significance. IL-17 is one of the main triggers of the proinflammatory response that could potentially be associated with early brain injury (global ischemia), vasospasm, delayed cerebral ischemia, and increased mortality. IL-17 quantification could be an early prognostic biomarker with clinical value [71]. IL-17 is more closely associated with neutrophil recruitment and activation among the various cytokines. The inhibition of RAR-related orphan receptor gamma T (RoRγt), the master transcription factor of IL-17, decreases the CSF recruitment of neutrophils and could be a therapeutic target to ameliorate DCI [72].
- -
- The levels of IL-1β, IL-18, and TNF-α in the CSF were elevated in aSAH patients and were positively associated with cerebral edema and acute hydrocephalus. CSF inflammatory cytokines might be useful biomarkers to assess the severity and predict outcomes [73].
3.4. Chemokines in CSF
3.5. Leucocytes and Monocytes in CSF
3.6. Endothelin-1 e NO Synthase
3.7. Excitotoxicity
3.8. Ostopontin and Matricellular Protein
3.9. Blood-Brain Barrier Disruption
3.10. Microglia M1 and M2 Polarization
3.11. Neuronal Apoptosis
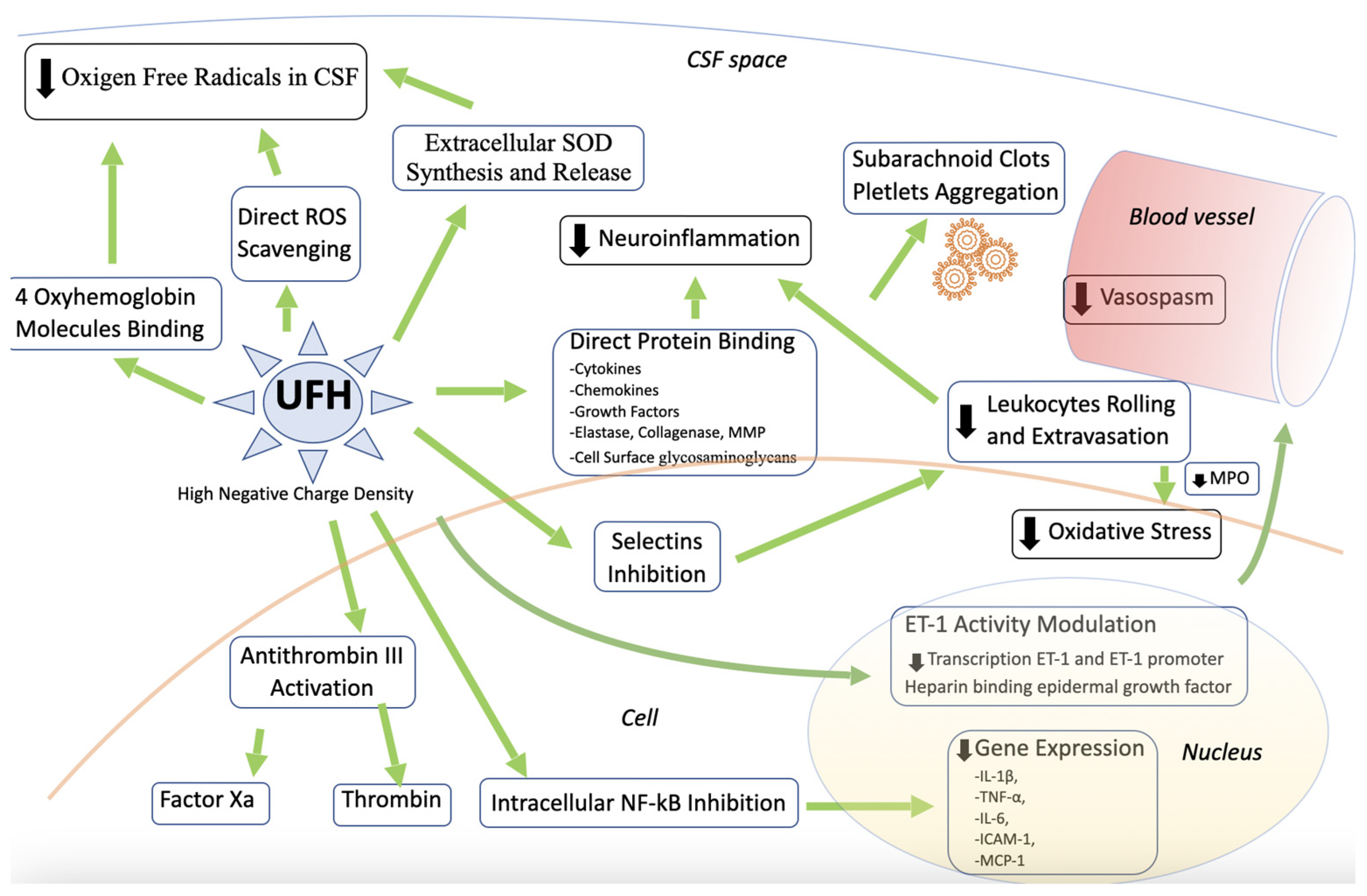
3.12. Heparin and SAH
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SAH | Subarachnoid hemorrhage |
| EBI | Early brain injury |
| DCI | Delayed cerebral ischemia |
| SSE | Subarachnoid Secondary Events |
| CSF | Cerebrospinal Fluid |
| ROS | Reactive Oxygen Species |
| NO | Nitric Oxide |
References
- Rincon, F.; Rossenwasser, R.H.; Dumont, A. The Epidemiology of Admissions of Nontraumatic Subarachnoid Hemorrhage in the United States. Neurosurgery 2013, 73, 217–222, discussion 212–213. [Google Scholar] [CrossRef] [PubMed]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and Functional Outcome After Aneurysmal Subarachnoid Hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kanamaru, H.; Kawakita, F.; Asada, R.; Fujimoto, M.; Shiba, M. Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol. Histopathol. 2021, 36, 143–158. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Colohan, A.R.; Zhang, J.H. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 399–414. [Google Scholar] [CrossRef]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2013, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.M.; Nisar, J.; Darsaut, T. Cerebral Vasospasm: A Review. Can. J. Neurol. Sci. 2015, 43, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Turan, N.; Chau, M.; Pradilla, G. Inflammation, Vasospasm, and Brain Injury after Subarachnoid Hemorrhage. BioMed Res. Int. 2014, 2014, 384342. [Google Scholar] [CrossRef]
- Geraghty, J.R.; Testai, F.D. Delayed Cerebral Ischemia after Subarachnoid Hemorrhage: Beyond Vasospasm and Towards a Multifactorial Pathophysiology. Curr. Atheroscler. Rep. 2017, 19, 50. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011, 10, 618–625. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Manoel, A.L.; Macdonald, R.L. Neuroinflammation as a Target for Intervention in Subarachnoid Hemorrhage. Front. Neurol. 2018, 9, 292. [Google Scholar] [CrossRef]
- Liu, G.J.; Luo, J.; Zhang, L.P.; Wang, Z.J.; Xu, L.L.; He, G.H.; Zeng, Y.J.; Wang, Y.F. Meta-analysis of the effectiveness and safety of prophylactic use of nimodipine in patients with an aneurysmal subarachnoid haemorrhage. CNS Neurol. Disord. Drug Targets 2011, 10, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Aldrich, E.F.; Schreibman, D.; James, R.F.; Polifka, A.; Beaty, N. Low-dose intravenous heparin infusion in patients with aneurysmal subarachnoid hemorrhage: A preliminary assessment. J. Neurosurg. 2013, 119, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Bruder, M.; Won, S.-Y.; Kashefiolasl, S.; Wagner, M.; Brawanski, N.; Dinc, N.; Seifert, V.; Konczalla, J. Effect of heparin on secondary brain injury in patients with subarachnoid hemorrhage: An additional ‘H’ therapy in vasospasm treatment. J. NeuroInterv. Surg. 2017, 9, 659–663. [Google Scholar] [CrossRef]
- James, R.F.; Khattar, N.K.; Aljuboori, Z.S.; Page, P.S.; Shao, E.Y.; Carter, L.M.; Meyer, K.S.; Daniels, M.W.; Craycroft, J.; Gaughen, J.R.; et al. Continuous infusion of low-dose unfractionated heparin after aneurysmal subarachnoid hemorrhage: A preliminary study of cognitive outcomes. J. Neurosurg. 2018, 130, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Kole, M.J.; Wessell, A.P.; Ugiliweneza, B.; Cannarsa, G.J.; Fortuny, E.; Stokum, J.A.; Shea, P.; Chryssikos, T.; Khattar, N.K.; Crabill, G.A.; et al. Low-Dose Intravenous Heparin Infusion After Aneurysmal Subarachnoid Hemorrhage is Associated With Decreased Risk of Delayed Neurological Deficit and Cerebral Infarction. Neurosurgery 2021, 88, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Patel, A.P.; James, R.F.; Simard, J.M. Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease. Molecules 2017, 22, 724. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Surg. 2021, 372, n71. [Google Scholar] [CrossRef]
- Pluta, R.M.; Afshar, J.K.B.; Boock, R.J.; Oldfield, E.H. Temporal changes in perivascular concentrations of oxyhemoglobin, deoxyhemoglobin, and methemoglobin after subarachnoid hemorrhage. J. Neurosurg. 1998, 88, 557–561. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Weir, B.K. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke 1991, 22, 971–982. [Google Scholar] [CrossRef]
- Hugelshofer, M.; Buzzi, R.M.; Schaer, C.; Richter, H.; Akeret, K.; Anagnostakou, V.; Mahmoudi, L.; Vaccani, R.; Vallelian, F.; Deuel, J.W.; et al. Haptoglobin administration into the subarachnoid space prevents hemoglobin-induced cerebral vasospasm. J. Clin. Investig. 2019, 129, 5219–5235. [Google Scholar] [CrossRef]
- Garland, P.; Morton, M.J.; Haskins, W.; Zolnourian, A.; Durnford, A.; Gaastra, B.; Toombs, J.; Heslegrave, A.J.; More, J.; Okemefuna, A.I.; et al. Haemoglobin causes neuronal damage in vivo which is preventable by haptoglobin. Brain Commun. 2020, 2, fcz053. [Google Scholar] [CrossRef]
- Buehler, P.W.; Humar, R.; Schaer, D.J. Haptoglobin Therapeutics and Compartmentalization of Cell-Free Hemoglobin Toxicity. Trends Mol. Med. 2020, 26, 683–697. [Google Scholar] [CrossRef]
- Akeret, K.; Buzzi, R.M.; Schaer, C.A.; Thomson, B.R.; Vallelian, F.; Wang, S.; Willms, J.; Sebök, M.; Held, U.; Deuel, J.W.; et al. Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury. J. Cereb. Blood Flow Metab. 2021, 41, 3000–3015. [Google Scholar] [CrossRef]
- Sabri, M.; Ai, J.; Lakovic, K.; D’abbondanza, J.; Ilodigwe, D.; Macdonald, R. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience 2012, 224, 26–37. [Google Scholar] [CrossRef]
- Radziwon-Balicka, A.; Lesyk, G.; Back, V.; Fong, T.; Loredo-Calderon, E.L.; Dong, B.; El-Sikhry, H.; El-Sherbeni, A.A.; El-Kadi, A.; Ogg, S.; et al. Differential eNOS-signalling by platelet subpopulations regulates adhesion and aggregation. Cardiovasc. Res. 2017, 113, 1719–1731. [Google Scholar] [CrossRef] [PubMed]
- Atochin, D.N.; Huang, P.L. Role of Endothelial Nitric Oxide in Cerebrovascular Regulation. Curr. Pharm. Biotechnol. 2011, 12, 1334–1342. [Google Scholar] [CrossRef]
- Hugelshofer, M.; Sikorski, C.M.; Seule, M.; Deuel, J.; Muroi, C.I.; Seboek, M.; Akeret, K.; Buzzi, R.; Regli, L.; Schaer, D.J.; et al. Cell-Free Oxyhemoglobin in Cerebrospinal Fluid After Aneurysmal Subarachnoid Hemorrhage: Biomarker and Potential Therapeutic Target. World Neurosurg. 2018, 120, e660–e666. [Google Scholar] [CrossRef]
- Thomas, A.J.; Ogilvy, C.S.; Griessenauer, C.J.; Hanafy, K.A. Macrophage CD163 expression in cerebrospinal fluid: Association with subarachnoid hemorrhage outcome. J. Neurosurg. 2018, 131, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Wu, F.; Liu, Z.; Li, G.; Zhou, L.; Huang, K.; Wu, Z.; Zhan, R.; Shen, J. Inflammation and Oxidative Stress: Potential Targets for Improving Prognosis After Subarachnoid Hemorrhage. Front. Cell. Neurosci. 2021, 15, 739506. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Bederson, J.B. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 381–398. [Google Scholar] [CrossRef]
- Chen, S.; Feng, H.; Sherchan, P.; Klebe, D.; Zhao, G.; Sun, X.; Zhang, J.; Tang, J.; Zhang, J.H. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog. Neurobiol. 2014, 115, 64–91. [Google Scholar] [CrossRef]
- Hao, G.; Eser, P.; Mo, J. Oxidative Stress and Intracranial Hypertension after Aneurysmal Subarachnoid Hemorrhage. Antioxidants 2022, 11, 2423. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, S.; Zhang, J.-M. The Updated Role of Oxidative Stress in Subarachnoid Hemorrhage. Curr. Drug DelCurr. Drug Deliv. 2017, 14, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Li, R.; Tu, W.-J.; Chen, Y.; Wang, K.; Chen, X.; Zhao, J. An Update on Antioxidative Stress Therapy Research for Early Brain Injury After Subarachnoid Hemorrhage. Front. Aging Neurosci. 2021, 13, 772036. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, A.; Liu, Y.; Hu, X.; Fang, Y.; Wang, X.; Luo, Y.; Lenahan, C.; Chen, S. New Mechanisms and Targets of Subarachnoid Hemorrhage: A Focus on Mitochondria. Curr. Neuropharmacol. 2022, 20, 1278–1296. [Google Scholar] [CrossRef]
- Krenzlin, H.; Wesp, D.; Schmitt, J.; Frenz, C.; Kurz, E.; Masomi-Bornwasser, J.; Lotz, J.; Ringel, F.; Kerz, T.; Keric, N. Decreased Superoxide Dismutase Concentrations (SOD) in Plasma and CSF and Increased Circulating Total Antioxidant Capacity (TAC) Are Associated with Unfavorable Neurological Outcome after Aneurysmal Subarachnoid Hemorrhage. J. Clin. Med. 2021, 10, 1188. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.V.; Suggs, J.M.; Diwan, D.; Lee, J.V.; Lipsey, K.; Vellimana, A.K.; Zipfel, G.J. Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: A review and synthesis. J. Cereb. Blood Flow Metab. 2020, 40, 1565–1575. [Google Scholar] [CrossRef]
- Dienel, A.; Kumar, T.P.; Blackburn, S.L.; McBride, D.W. Role of platelets in the pathogenesis of delayed injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2021, 41, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Morel, N.; Freyssinet, J.-M.; Toti, F. Platelet microparticles and vascular cells interactions: A checkpoint between the haemostatic and thrombotic responses. Platelets 2008, 19, 9–23. [Google Scholar] [CrossRef]
- Perez, P.; Lukaszewicz, A.-C.; Lenck, S.; Nizard, R.; Drouet, L.; Payen, D. Platelet activation and aggregation after aneurysmal subarachnoid hemorrhage. BMC Neurol. 2018, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kusaka, G.; Yamaguchi, N.; Sekizuka, E.; Nakadate, H.; Minamitani, H.; Shinoda, S.; Watanabe, E. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery 2009, 64, 546–554, discussion 553–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-W.; Yanamoto, H.; Nagata, I.; Miyamoto, S.; Nakajo, Y.; Xue, J.-H.; Iihara, K.; Kikuchi, H. Platelet-Derived Growth Factor-Induced Severe and Chronic Vasoconstriction of Cerebral Arteries. Neurosurgery 2010, 66, 728–735, discussion 735. [Google Scholar] [CrossRef]
- Gaetani, P.; Tancioni, F.; Grignani, G.; Tartara, F.; Merlo, E.M.; Brocchieri, A.; Rodriguez, Y.; Baena, R. Platelet derived growth factor and subarachnoid haemorrage: A study on cisternal cerebrospinal fluid. Acta Neurochir. 1997, 139, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ghali, M.G.Z.; Srinivasan, V.M.; Johnson, J.; Kan, P.; Britz, G. Therapeutically Targeting Platelet-Derived Growth Fac-tor-Mediated Signaling Underlying the Pathogenesis of Subarachnoid Hemorrhage-Related Vasospasm. J. Stroke Cerebrovasc. Dis. 2018, 27, 2289–2295. [Google Scholar] [CrossRef]
- Mastellos, D.; Morikis, D.; Isaacs, S.N.; Holland, M.C.; Strey, C.W.; Lambris, J.D. Complement: Structure, Functions, Evolution, and Viral Molecular Mimicry. Immunol. Res. 2003, 27, 367–386. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, B.J.; Meijers, J.C.M.; Kloek, A.T.; Knaup, V.L.; Rinkel, G.J.E.; Morgan, B.P.; van der Kamp, M.J.; Osuka, K.; Aronica, E.; Ruigrok, Y.M.; et al. Complement C5 Contributes to Brain Injury After Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 11, 678–688. [Google Scholar] [CrossRef]
- Koopman, I.; Rinkel, G.J.E.; Vergouwen, M.D.I.; CLASH Study Group. CompLement C5 Antibodies for decreasing brain injury after aneurysmal Subarachnoid Haemorrhage (CLASH): Study protocol for a randomised controlled phase II clinical trial. Trials 2020, 21, 969. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Shin, M.L.; Simard, J.M. The complement membrane attack complex and the bystander effect in cerebral vasospasm. J. Neurosurg. 1997, 87, 294–300. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed]
- Llull, L.; Thiel, S.; Amaro, S.; Cervera, Á.; Planas, A.M.; Chamorro, Á. Ficolin-1 Levels in Patients Developing Vasospasm and Cerebral Ischemia After Spontaneous Subarachnoid Hemorrhage. Mol. Neurobiol. 2016, 54, 6572–6580. [Google Scholar] [CrossRef]
- Sandgaard, E.; Troldborg, A.; Lauridsen, S.V.; Gyldenholm, T.; Thiel, S.; Hvas, A.-M. Changes in the Lectin Pathway Following Intracerebral or Spontaneous Subarachnoid Hemorrhage. Mol. Neurobiol. 2018, 56, 78–87. [Google Scholar] [CrossRef]
- Zanier, E.R.; Zangari, R.; Munthe-Fog, L.; Hein, E.; Zoerle, T.; Conte, V.; Orsini, F.; Tettamanti, M.; Stocchetti, N.; Garred, P.; et al. Ficolin-3-mediated lectin complement pathway activation in patients with subarachnoid hemorrhage. Neurology 2014, 82, 126–134. [Google Scholar] [CrossRef]
- Cai, J.-Y.; Sun, J.; Yu, Z.-Q. Serum mannose-binding lectin levels after aneurysmal subarachnoid hemorrhage. Acta Neurol. Scand. 2016, 134, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Matzen, J.S.; Krogh, C.L.; Forman, J.L.; Garred, P.; Møller, K.; Bache, S. Lectin complement pathway initiators after subarachnoid hemorrhage—An observational study. J. Neuroinflamm. 2020, 17, 338. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, P.; Tartara, F.; Pignattit, P.; Tancioni, F.; Rodriguez, R.; Baena, B.; De Benedettit, F. Cisternal CSF levels of cytokines after subarachnoid hemorrhage. Neurol. Res. 1998, 20, 337–342. [Google Scholar] [CrossRef]
- Ridwan, S.; Grote, A.; Simon, M. Interleukin 6 in cerebrospinal fluid is a biomarker for delayed cerebral ischemia (DCI) related infarctions after aneurysmal subarachnoid hemorrhage. Sci. Rep. 2021, 11, 12. [Google Scholar] [CrossRef]
- Ďuriš, K.; Neuman, E.; Vybíhal, V.; Juráň, V.; Gottwaldová, J.; Kýr, M.; Vašků, A.; Smrčka, M. Early Dynamics of Interleukin-6 in Cerebrospinal Fluid after Aneurysmal Subarachnoid Hemorrhage. J. Neurol. Surg. Part A: Central Eur. Neurosurg. 2017, 79, 145–151. [Google Scholar] [CrossRef]
- Ni, W.; Gu, Y.X.; Song, D.L.; Leng, B.; Li, P.L.; Mao, Y. The Relationship Between IL-6 in CSF and Occurrence of Vasospasm After Subarachnoid Hemorrhage. Acta Neurochir. Suppl. 2011, 110 Pt 1, 203–208. [Google Scholar] [CrossRef]
- Lenski, M.; Huge, V.; Briegel, J.; Tonn, J.-C.; Schichor, C.; Thon, N. Interleukin 6 in the Cerebrospinal Fluid as a Biomarker for Onset of Vasospasm and Ventriculitis After Severe Subarachnoid Hemorrhage. World Neurosurg. 2017, 99, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wostrack, M.; Reeb, T.; Martin, J.; Kehl, V.; Shiban, E.; Preuss, A.; Ringel, F.; Meyer, B.; Ryang, Y.-M. Shunt-Dependent Hydrocephalus After Aneurysmal Subarachnoid Hemorrhage: The Role of Intrathecal Interleukin-6. Neurocrit. Care 2014, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.M.; Sivanrupan, S.; Wanderer, S.; Agnoletto, G.J.; Chiappini, A.; Grüter, B.E.; Andereggen, L.; Mariani, L.; Taussky, P.; Marbacher, S. Preclinical and clinical role of interleukin-6 in the development of delayed cerebral vasospasm and neuronal cell death after subarachnoid hemorrhage: Towards a potential target therapy? Neurosurg. Rev. 2022, 45, 395–403. [Google Scholar] [CrossRef]
- Wu, W.; Guan, Y.; Zhao, G.; Fu, X.-J.; Guo, T.-Z.; Liu, Y.-T.; Ren, X.-L.; Wang, W.; Liu, H.-R.; Li, Y.-Q. Elevated IL-6 and TNF-α Levels in Cerebrospinal Fluid of Subarachnoid Hemorrhage Patients. Mol. Neurobiol. 2016, 53, 3277–3285. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Grobelny, B.; Fernandez, L.; Kurtz, P.; Connolly, E.; Mayer, S.A.; Schindler, C.; Badjatia, N. Brain interstitial fluid TNF-α after subarachnoid hemorrhage. J. Neurol. Sci. 2010, 291, 69–73. [Google Scholar] [CrossRef]
- Han, M.; Liu, D.; Qiu, J.; Yuan, H.; Hu, Q.; Xue, H.; Li, T.; Ma, W.; Zhang, Q.; Li, G.; et al. Evaluation of H2S-producing enzymes in cerebrospinal fluid and its relationship with interleukin-6 and neurologic deficits in subarachnoid hemorrhage. Biomed. Pharmacother. 2020, 123, 109722. [Google Scholar] [CrossRef]
- Douglas, M.R.; Daniel, M.; Lagord, C.; Akinwunmi, J.; Jackowski, A.; Cooper, C.; Berry, M.; Logan, A. High CSF transforming growth factor beta levels after subarachnoid haemorrhage: Association with chronic communicating hydrocephalus. J. Neurol. Neurosurg. Psychiatry 2009, 80, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Nakamura, T.; Shinomiya, A.; Kawakita, K.; Kawanishi, M.; Miyake, K.; Kuroda, Y.; Keep, R.F.; Tamiya, T. Histidine-rich Glycoprotein Could Be an Early Predictor of Vasospasm after Aneurysmal Subarachnoid Hemorrhage. Acta Med. Okayama 2019, 73, 29–39. [Google Scholar]
- Lad, S.P.; Hegen, H.; Gupta, G.; Deisenhammer, F.; Steinberg, G.K. Proteomic Biomarker Discovery in Cerebrospinal Fluid for Cerebral Vasospasm Following Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2012, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Trias, N.; Brugnini, A.; Grille, P.; Lens, D.; Biestro, A.; Grille, S. TH17/Treg imbalance and IL-17A increase after severe aneurysmal subarachnoid hemorrhage. J. Neuroimmunol. 2020, 346, 577310. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, A.P.; Gartman, W.T.; Swank, V.; Gomes, J.A.; Ruozhuo, L.; DeBacker, J.; Provencio, J.J. RAR-Related Orphan Receptor Gamma T (RoRγt)-Related Cytokines Play a Role in Neutrophil Infiltration of the Central Nervous System After Subarachnoid Hemorrhage. Neurocrit. Care 2020, 33, 140–151. [Google Scholar] [CrossRef]
- Lv, S.-Y.; Wu, Q.; Liu, J.-P.; Shao, J.; Wen, L.-L.; Xue, J.; Zhang, X.-S.; Zhang, Q.-R.; Zhang, X. Levels of Interleukin-1β, Interleukin-18, and Tumor Necrosis Factor-α in Cerebrospinal Fluid of Aneurysmal Subarachnoid Hemorrhage Patients May Be Predictors of Early Brain Injury and Clinical Prognosis. World Neurosurg. 2018, 111, e362–e373. [Google Scholar] [CrossRef]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef]
- Niwa, A.; Osuka, K.; Nakura, T.; Matsuo, N.; Watabe, T.; Takayasu, M. Interleukin-6, MCP-1, IP-10, and MIG are sequentially expressed in cerebrospinal fluid after subarachnoid hemorrhage. J. Neuroinflamm. 2016, 13, 217. [Google Scholar] [CrossRef]
- Lu, H.; Shi, J.-X.; Chen, H.-L.; Hang, C.-H.; Wang, H.-D.; Yin, H.-X. Expression of monocyte chemoattractant protein-1 in the cerebral artery after experimental subarachnoid hemorrhage. Brain Res. 2009, 1262, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Handa, Y.; Tsuchida, A.; Kaneko, M.; Kobayashi, H.; Kubota, T. The kinetics of lymphocyte subsets and macrophages in subarachnoid space after subarachnoid hemorrhage in rats. Stroke 1993, 24, 1993–2000, discussion 2000–2001. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.R.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Dobreva, G.; Hänggi, D.; Muhammad, S. Elevated level of cerebrospinal fluid and systemic chemokine CCL5 is a predictive biomarker of clinical outcome after aneurysmal subarachnoid hemorrhage (aSAH). Cytokine 2020, 133, 155142. [Google Scholar] [CrossRef]
- Levy, J.A. The Unexpected Pleiotropic Activities of RANTES. J. Immunol. 2009, 182, 3945–3946. [Google Scholar] [CrossRef]
- Rollins, B.J. Chemokines. Blood 1997, 90, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Sauvigny, T.; Mader, M.M.-D.; Schweingruber, N.; Maire, C.L.; Rünger, A.; Ricklefs, F.; Regelsberger, J.; Schmidt, N.O.; Westphal, M.; et al. Immune Characterization in Aneurysmal Subarachnoid Hemorrhage Reveals Distinct Monocytic Activation and Chemokine Patterns. Transl. Stroke Res. 2020, 11, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Xu, H.-L.; Garcia, M.; Testai, F.; Vetri, F.; Barabanova, A.; Pelligrino, D.A.; Paisansathan, C. Pharmacologic blockade of vascular adhesion protein-1 lessens neurologic dysfunction in rats subjected to subarachnoid hemorrhage. Brain Res. 2014, 1586, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Gris, T.; Laplante, P.; Thebault, P.; Cayrol, R.; Najjar, A.; Joannette-Pilon, B.; Brillant-Marquis, F.; Magro, E.; English, S.W.; Lapointe, R.; et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J. Neuroinflamm. 2019, 16, 253. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Fu, X.; Siu, A.; Rasmussen, P.A.; Hazen, S.L.; Ransohoff, R.M. CSF Neutrophils Are Implicated in the Development of Vasospasm in Subarachnoid Hemorrhage. Neurocrit. Care 2009, 12, 244–251. [Google Scholar] [CrossRef]
- Lim, M.; Bower, R.S.; Wang, Y.; Sims, L.; Bower, M.R.; Camara-Quintana, J.; Li, G.; Cheshier, S.; Harsh, G.R.; Steinberg, G.K.; et al. The predictive value of serum myeloperoxidase for vasospasm in patients with aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 2012, 35, 413–419. [Google Scholar] [CrossRef]
- Roa, J.A.; Sarkar, D.; Zanaty, M.; Ishii, D.; Lu, Y.; Karandikar, N.J.; Hasan, D.M.; Ortega, S.B.; Samaniego, E.A. Preliminary results in the analysis of the immune response after aneurysmal subarachnoid hemorrhage. Sci. Rep. 2020, 10, 11809. [Google Scholar] [CrossRef]
- Kaynar, M.Y.; Tanriverdi, T.; Kafadar, A.M.; Kacira, T.; Uzun, H.; Aydin, S.; Gumustas, K.; Dirican, A.; Kuday, C. Detection of soluble intercellular adhesion molecule—1 and vascular cell adhesion molecule—1 in both cerebrospinal fluid and serum of patients after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 1030–1036. [Google Scholar] [CrossRef]
- Polin, R.S.; Bavbek, M.; Shaffrey, M.E.; Billups, K.; Bogaev, C.A.; Kassell, N.F.; Lee, K.S. Detection of soluble E-selectin, ICAM-1, VCAM-1, and L-selectin in the cerebrospinal fluid of patients after subarachnoid hemorrhage. J. Neurosurg. 1998, 89, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, A.; Matsumoto, M.; Tedder, T.F.; Lowe, J.B.; Miyasaka, M.; Hirata, T. An L-selectin ligand distinct from P-selectin glycoprotein ligand-1 is expressed on endothelial cells and promotes neutrophil rolling in inflammation. Blood 2008, 112, 4915–4923. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Grille, S.; Morelli, P.; Mila, R.; Trias, N.; Brugnini, A.; Lluberas, N.; Biestro, A.; Lens, D. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with Aneurysmal Subarachnoid Hemorrhage. Springerplus 2015, 4, 195. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerg. 2010, 2, 5. [Google Scholar] [CrossRef]
- Yeung, P.K.; Shen, J.; Chung, S.S.; Chung, S.K. Targeted over-expression of endothelin-1 in astrocytes leads to more severe brain damage and vasospasm after subarachnoid hemorrhage. BMC Neurosci. 2013, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Kawanabe, Y.; Nauli, S.M. Endothelin. Cell. Mol. Life Sci. 2011, 68, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kessler, I.M.; Pacheco, Y.G.; Lozzi, S.P.; de Araújo, A.S., Jr.; Onishi, F.J.; de Mello, P.A. Endothelin-1 levels in plasma and cerebrospinal fluid of patients with cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Surg. Neurol. 2005, 64 (Suppl. S1), S2–S5, discussion S1–S5. [Google Scholar] [CrossRef]
- Mascia, L.; Fedorko, L.; Stewart, D.J.; Mohamed, F.; Terbrugge, K.; Ranieri, V.M.; Wallace, M.C. Temporal Relationship Between Endothelin-1 Concentrations and Cerebral Vasospasm in Patients With Aneurysmal Subarachnoid Hemorrhage. Stroke 2001, 32, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Seifert, V.; Löffler, B.-M.; Zimmermann, M.; Roux, S.; Stolke, D. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage. Correlation with cerebral vasospasm, delayed ischemic neurological deficits, and volume of hematoma. J. Neurosurg. 1995, 82, 55–62. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, W.; Dou, X.; Jia, R.; Yang, H.; Liu, X.; Xu, C.; Liu, J.; Cao, Y.; Luo, G. Role of endothelin-1 and its receptors in cerebral vasospasm following subarachnoid hemorrhage. Mol. Med. Rep. 2018, 18, 5229–5236. [Google Scholar] [CrossRef]
- Suzuki, R.; Masaoka, H.; Hirata, Y.; Marumo, F.; Isotani, E.; Hirakawa, K. The role of endothelin-1 in the origin of cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 1992, 77, 96–100. [Google Scholar] [CrossRef]
- Wanebo, J.E.; Arthur, A.S.; Louis, H.G.; West, K.; Kassell, N.F.; Lee, K.S.; Helm, G.A. Systemic administration of the endothelin-A receptor antagonist TBC 11251 attenuates cerebral vasospasm after experimental subarachnoid hemorrhage: Dose study and review of endothelin-based therapies in the literature on cerebral vasospasm. Neurosurgery 1998, 43, 1409–1417, discussion 1417–1418. [Google Scholar] [CrossRef]
- Ng, W.H.; Moochhala, S.; Yeo, T.T.; Ong, P.L.; Ng, P.Y. Nitric Oxide and Subarachnoid Hemorrhage: Elevated Levels in Cerebrospinal Fluid and Their Implications. Neurosurgery 2001, 49, 622–627, discussion 626–627. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Li, J.; Zhao, Z.; Gao, C.; Sun, Z.; Liu, X. Effects of tetramethylpyrazine on nitric oxide/cGMP signaling after cerebral vasospasm in rabbits. Brain Res. 2010, 1361, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.S.; Iuliano, B.A.; Harvey-White, J.; Espey, M.G.; Oldfield, E.H.; Pluta, R.M. Association between cerebrospinal fluid levels of asymmetric dimethyl-L-arginine, an endogenous inhibitor of endothelial nitric oxide synthase, and cerebral vasospasm in a primate model of subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 836–842. [Google Scholar] [CrossRef]
- Woszczyk, A.; Deinsberger, W. Nitric oxide metabolites in cisternal CSF correlate with cerebral vasospasm in patients with a subarachnoid haemorrhage. Acta Neurochir. 2003, 145, 257–264, discussion 263–264. [Google Scholar] [CrossRef] [PubMed]
- Seng, K.G.; Kandasamy, R.; Bujang, M.A.; Swammy, M.; Mustapha, M.; Abdullah, J.M. Ratio of Nitric Oxide Metabolite Levels in Cerebrospinal Fluid and Serum, and Their Correlation with Severity and Outcome in Patients with Subarachnoid Haemorrhage. Malays. J. Med. Sci. 2021, 28, 42–54. [Google Scholar] [CrossRef]
- Pluta, R.M. Nitrite Infusions to Prevent Delayed Cerebral Vasospasm in a Primate Model of Subarachnoid Hemorrhage. JAMA 2005, 293, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.M. Delayed cerebral vasospasm and nitric oxide: Review, new hypothesis, and proposed treatment. Pharmacol. Ther. 2005, 105, 23–56. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Suzuki, H.; Kawakita, F.; Asada, R.; Nakano, F.; Nishikawa, H.; Fujimoto, M. Old but still hot target, gluta-mate-mediated neurotoxicity in stroke. Transl Stroke Res. 2022, 13, 216–217. [Google Scholar] [CrossRef]
- Zimmermann, J.; Weller, J.; Grub, S.; Kebir, S.; Lehmann, F.; Vatter, H.; Schuss, P.; Güresir, E.; Müller, M. Arginase-1 Released into CSF After Aneurysmal Subarachnoid Hemorrhage Decreases Arginine/Ornithine Ratio: A Novel Prognostic Biomarker. Transl. Stroke Res. 2022, 13, 382–390. [Google Scholar] [CrossRef]
- Ruppel, R.A.; Kochanek, P.M.; Adelson, P.; Rose, M.E.; Wisniewski, S.; Bell, M.J.; Clark, R.S.; Marion, D.W.; Graham, S.H. Excitatory amino acid concentrations in ventricular cerebrospinal fluid after severe traumatic brain injury in infants and children: The role of child abuse. J. Pediatr. 2001, 138, 18–25. [Google Scholar] [CrossRef]
- Sokół, B.; Urbaniak, B.; Wąsik, N.; Plewa, S.; Klupczynska, A.; Jankowski, R.; Więckowska, B.; Juszkat, R.; Kokot, Z. Amino Acids in Cerebrospinal Fluid of Patients with Aneurysmal Subarachnoid Haemorrhage: An Observational Study. Front. Neurol. 2017, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Bowen, D.M.; DeKosky, S.T. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 1994, 6, 153–156. [Google Scholar] [CrossRef]
- Suzuki, H.; Kawakita, F.; Asada, R. Neuroelectric Mechanisms of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2022, 23, 3102. [Google Scholar] [CrossRef]
- Kawakita, F.; Kanamaru, H.; Asada, R.; Suzuki, Y.; Nampei, M.; Nakajima, H.; Oinaka, H.; Suzuki, H. Roles of glutamate in brain injuries after subarachnoid hemorrhage. Histol. Histopathol. 2022, 37, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.; Olivares-Rivera, A.; Major, S.; Sánchez-Porras, R.; Uhlmann, L.; Kunzmann, K.; Zerelles, R.; Kentar, M.; Kola, V.; Aguilera, A.H.; et al. Lasting s-ketamine block of spreading depolarizations in subarachnoid hemorrhage: A retrospective cohort study. Crit. Care 2019, 23, 427. [Google Scholar] [CrossRef]
- Abate, M.G.; Moretto, L.; Licari, I.; Esposito, T.; Capuano, L.; Olivieri, C.; Benech, A.; Brucoli, M.; Avanzi, G.C.; Cammarota, G.; et al. Osteopontin in the Cerebrospinal Fluid of Patients with Severe Aneurysmal Subarachnoid Hemorrhage. Cells 2019, 8, 695. [Google Scholar] [CrossRef]
- Rosmus, D.-D.; Lange, C.; Ludwig, F.; Ajami, B.; Wieghofer, P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines 2022, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hasegawa, Y.; Chen, W.; Kanamaru, K.; Zhang, J.H. Recombinant osteopontin in cerebral vasospasm after subarachnoid hemorrhage. Ann. Neurol. 2010, 68, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Asada, R.; Suzuki, H. Osteopontin in post-subarachnoid hemorrhage pathologies. J. Integr. Neurosci. 2022, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Asada, R.; Nakatsuka, Y.; Kanamaru, H.; Kawakita, F.; Fujimoto, M.; Miura, Y.; Shiba, M.; Yasuda, R.; Toma, N.; Suzuki, H.; et al. Higher Plasma Osteopontin Concentrations Associated with Subsequent Development of Chronic Shunt-Dependent Hydrocephalus After Aneurysmal Subarachnoid Hemorrhage. Transl. Stroke Res. 2021, 12, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Solár, P.; Zamani, A.; Lakatosová, K.; Joukal, M. The blood–brain barrier and the neurovascular unit in subarachnoid hemorrhage: Molecular events and potential treatments. Fluids Barriers CNS 2022, 19, 29. [Google Scholar] [CrossRef]
- Marbacher, S.; Andereggen, L.; Neuschmelting, V.; Widmer, H.R.; von Gunten, M.; Takala, J.; Jakob, S.M.; Fandino, J. A new rabbit model for the study of early brain injury after subarachnoid hemorrhage. J. Neurosci. Methods 2012, 208, 138–145. [Google Scholar] [CrossRef]
- Kumar, T.P.; McBride, D.W.; Dash, P.K.; Matsumura, K.; Rubi, A.; Blackburn, S.L. Endothelial Cell Dysfunction and Injury in Subarachnoid Hemorrhage. Mol. Neurobiol. 2019, 56, 1992–2006. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ostrowski, R.P.; Sun, X.; Ma, Q.; Tang, J.; Zhang, J.H. Targeting C/EBP homologous protein with siRNA attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. Exp. Neurol. 2012, 238, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yamaguchi, M.; Kusaka, G.; Schonholz, C.; Nanda, A.; Zhang, J.H. Caspase Inhibitors Prevent Endothelial Apoptosis and Cerebral Vasospasm in Dog Model of Experimental Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2004, 24, 419–431. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35 Pt 5, 1147–1150. [Google Scholar] [CrossRef]
- Frei, B. Reactive oxygen species and antioxidant vitamins: Mechanisms of action. Am. J. Med. 1994, 97, S5–S13, discussion S22–S28. [Google Scholar] [CrossRef]
- Sen, O.; Caner, H.; Aydin, M.V.; Ozen, O.; Atalay, B.; Altınörs, N.; Bavbek, M. The effect of mexiletine on the level of lipid peroxidation and apoptosis of endothelium following experimental subarachnoid hemorrhage. Neurol. Res. 2006, 28, 859–863. [Google Scholar] [CrossRef]
- Friedrich, V.; Flores, R.; Muller, A.; Bi, W.; Peerschke, E.I.; Sehba, F.A. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. J. Neuroinflam. 2011, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Mostafa, G.; Knopman, J.; Friedrich, V., Jr.; Bederson, J.B. Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 633–640, discussion 219–220. [Google Scholar] [CrossRef]
- Zubkov, A.Y.; Tibbs, R.E.; Aoki, K.; Zhang, J.H. Morphological changes of cerebral penetrating arteries in a canine double hemorrhage model. Surg. Neurol. 2000, 54, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Chen, C.; Hu, Q.; Yang, X.; Lei, J.; Yang, L.; Wang, K.; Qin, L.; Huang, H.; Zhou, C. The role of p53 in brain edema after 24 h of experimental subarachnoid hemorrhage in a rat model. Exp. Neurol. 2008, 214, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Wessell, A.; Gerzanich, V.; Sheth, K.N.; Simard, J.M. Mechanisms of Global Cerebral Edema Formation in Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care 2017, 26, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.-C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef]
- Zheng, Z.V.; Lyu, H.; Lam, S.Y.E.; Lam, P.K.; Poon, W.S.; Wong, G.K.C. The Dynamics of Microglial Polarization Reveal the Resident Neuroinflammatory Responses After Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 11, 433–449. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Watabe, K. The roles of microglia macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017, 22, 1805–1829. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, B.; Li, Y.; Zhang, Y.; Shao, J.; Wu, P.; Xu, C.; Chen, G.; Shi, H. Activation of RARα Receptor Attenuates Neuroinflammation After SAH via Promoting M1-to-M2 Phenotypic Polarization of Microglia and Regulating Mafb/Msr1/PI3K-Akt/NF-κB Pathway. Front. Immunol. 2022, 13, 839796. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.V.; Chen, J.; Lyu, H.; Lam, S.Y.E.; Lu, G.; Chan, W.Y.; Wong, G.K.C. Novel role of STAT3 in microglia-dependent neuroinflammation after experimental subarachnoid haemorrhage. Stroke Vasc. Neurol. 2022, 7, 62–70. [Google Scholar] [CrossRef]
- Wei, S.; Luo, C.; Yu, S.; Gao, J.; Liu, C.; Wei, Z.; Zhang, Z.; Wei, L.; Yi, B. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp. Cell Res. 2017, 361, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Cheyuo, C.; Jacob, A.; Wu, R.; Zhou, M.; Qi, L.; Dong, W.; Ji, Y.; Chaung, W.W.; Wang, H.; Nicastro, J.; et al. Recombinant human MFG-E8 attenuates cerebral ischemic injury: Its role in anti-inflammation and anti-apoptosis. Neuropharmacology 2012, 62, 890–900. [Google Scholar] [CrossRef]
- Gao, Y.-Y.; Tao, T.; Wu, D.; Zhuang, Z.; Lu, Y.; Wu, L.-Y.; Liu, G.-J.; Zhou, Y.; Zhang, D.-D.; Wang, H.; et al. MFG-E8 attenuates inflammation in subarachnoid hemorrhage by driving microglial M2 polarization. Exp. Neurol. 2021, 336, 113532. [Google Scholar] [CrossRef]
- Prunell, G.F.; Svendgaard, N.-A.; Alkass, K.; Mathiesen, T. Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J. Neurosurg. 2005, 102, 1046–1054. [Google Scholar] [CrossRef]
- Simard, J.M.; Schreibman, D.; Aldrich, E.F.; Stallmeyer, B.; Le, B.; James, R.F.; Beaty, N. Unfractionated Heparin: Multitargeted Therapy for Delayed Neurological Deficits Induced by Subarachnoid Hemorrhage. Neurocrit. Care 2010, 13, 439–449. [Google Scholar] [CrossRef]
- James, R.F.; Kramer, D.R.; Aljuboori, Z.; Parikh, G.; Adams, S.W.; Eaton, J.C.; Al-Shaar, H.A.; Badjatia, N.; Mack, W.J.; Simard, J.M. Novel Treatments in Neuroprotection for Aneurysmal Subarachnoid Hemorrhage. Curr. Treat. Options Neurol. 2016, 18, 38. [Google Scholar] [CrossRef]
- Lukito, P.P.; Lie, H.; Helsa, K.; July, J. Heparin in the treatment of aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. Neurosurg. Focus 2022, 52, E9. [Google Scholar] [CrossRef] [PubMed]
- Wurm, G.; Tomancok, B.; Nussbaumer, K.; Adelwöhrer, C.; Holl, K. Reduction of ischemic sequelae following spontaneous subarachnoid hemorrhage: A double-blind, randomized comparison of enoxaparin versus placebo. Clin. Neurol. Neurosurg. 2004, 106, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Kandrotas, R.J. Heparin Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 1992, 22, 359–374. [Google Scholar] [CrossRef]
- Gray, E.; Mulloy, B.; Barrowcliffe, T.W. Heparin and low-molecular-weight heparin. Thromb. Haemost. 2008, 99, 807–818. [Google Scholar] [CrossRef]
- Young, E. The anti-inflammatory effects of heparin and related compounds. Thromb. Res. 2008, 122, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Moradi, M.; Khorshidahmad, T.; Motamedi, M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv. Pharmacol. Sci. 2015, 2015, 507151. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef]
- Najjam, S.; Gibbs, R.V.; Gordon, M.Y.; Rider, C.C. Characterization of human recombinant interleukin 2 binding to heparin and heparan sulfate using an elisa approach. Cytokine 1997, 9, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Najjam, S.; Gordon, M.Y.; Gibbs, R.V.; Rider, C.C. IL-12 is a heparin-binding cytokine. J. Immunol. 1999, 162, 1064–1070. [Google Scholar] [CrossRef]
- Nagata, K.; Browne, K.D.; Suto, Y.; Kumasaka, K.; Cognetti, J.; Johnson, V.E.; Marks, J.; Smith, D.H.; Pascual, J. Early heparin administration after traumatic brain injury. J. Trauma Inj. Infect. Crit. Care 2017, 83, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.H.; Seo, G.H.; Lee, J.; Kim, J.H.; Kim, D.G.; Ahn, Y.S. Heparin Attenuates the Expression of TNFα-induced Cerebral Endothelial Cell Adhesion Molecule. Korean J. Physiol. Pharmacol. 2008, 12, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Norgard-Sumnicht, K.; Linhardt, R.; Varki, A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J. Clin. Investig. 1998, 101, 877–889. [Google Scholar] [CrossRef]
- Nakane, H.; Chu, Y.; Faraci, F.; Oberley, L.W.; Heistad, D.D. Gene Transfer of Extracellular Superoxide Dismutase Increases Superoxide Dismutase Activity in Cerebrospinal Fluid. Stroke 2001, 32, 184–189. [Google Scholar] [CrossRef]
- Simard, J.M.; Tosun, C.; Ivanova, S.; Kurland, D.B.; Hong, C.; Radecki, L.; Gisriel, C.; Mehta, R.; Schreibman, D.; Gerzanich, V. Heparin Reduces Neuroinflammation and Transsynaptic Neuronal Apoptosis in a Model of Subarachnoid Hemorrhage. Transl. Stroke Res. 2012, 3 (Suppl. S1), 155–165. [Google Scholar] [CrossRef]
- Nagata, K.; Kumasaka, K.; Browne, K.D.; Li, S.; St-Pierre, J.; Cognetti, J.; Marks, J.; Johnson, V.E.; Smith, D.H.; Pascual, J.L. Unfractionated heparin after TBI reduces in vivo cerebrovascular inflammation, brain edema and accelerates cognitive recovery. J. Trauma Inj. Infect. Crit. Care 2016, 81, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Amiconi, G.; Zolla, L.; Vecchini, P.; Brunori, M.; Antonini, E. The Effect of Macromolecular Polyanions on the Functional Properties of Human Hemoglobin. Eur. J. Biochem. 1977, 76, 339–343. [Google Scholar] [CrossRef]
- Ross, M.A.; Long, W.F.; Williamson, F.B. Inhibition by heparin of Fe(II)-catalysed free-radical peroxidation of linolenic acid. Biochem. J. 1992, 286 Pt 3, 717–720. [Google Scholar] [CrossRef]
- Albertini, R.; Rindi, S.; Passi, A.; Pallavicini, G.; De Luca, G. Heparin protection against Fe2+-and Cu2+-mediated oxidation of liposomes. FEBS Lett. 1996, 383, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Yamada, H.; Futenma, A.; Kato, K.; Hirano, K. Heparin-Induced Release of Extracellular-Superoxide Dismutase Form (V) to Plasma. J. Biochem. 1995, 117, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Sandström, J.; Nilsson, P.; Karlsson, K.; Marklund, S.L. 10-fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. J. Biol. Chem. 1994, 269, 19163–19166. [Google Scholar] [CrossRef]
- Adachi, T.; Hara, H.; Yamada, H.; Yamazaki, N.; Yamamoto, M.; Sugiyama, T.; Futenma, A.; Katagiri, Y. Heparin-stimulated expression of extracellular-superoxide dismutase in human fibroblasts. Atherosclerosis 2001, 159, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.R.; Angstwurm, K.; Rosenkranz, T.; Lindauer, U.; Freyer, D.; Bürger, W.; Busch, C.; Einhäupl, K.M.; Dirnagl, U. Heparin Inhibits Leukocyte Rolling in Pial Vessels and Attenuates Inflammatory Changes in a Rat Model of Experimental Bacterial Meningitis. J. Cereb. Blood Flow Metab. 1997, 17, 1221–1229. [Google Scholar] [CrossRef]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara-Watanabe, K.; Hidai, C.; Ikeda, H.; Aoka, Y.; Ichikawa, K.-I.; Iguchi, N.; Okada-Ohno, M.; Yokota, J.; Kasanuki, H.; Kawana, M. Heparin Regulates Transcription of Endothelin-1 Gene in Endothelial Cells. J. Vasc. Res. 2005, 42, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chansel, D.; Ciroldi, M.; Vandermeersch, S.; Jackson, L.F.; Gomez, A.; Henrion, D.; Lee, D.C.; Coffman, T.M.; Richard, S.; Dussaule, J.; et al. Heparin binding EGF is necessary for vasospastic response to endothelin. FASEB J. 2006, 20, 1936–1938. [Google Scholar] [CrossRef]
- Kalmes, A.; Vesti, B.R.; Daum, G.; Abraham, J.A.; Clowes, A.W. Heparin blockade of thrombin-induced smooth muscle cell migration involves inhibition of epidermal growth factor (EGF) receptor transactivation by heparin-binding EGF-like growth factor. Circ. Res. 2000, 87, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zuo, G.; Shi, X.-Y.; Zhang, J.; Fang, Q.; Chen, G. Progesterone Administration Modulates Cortical TLR4/NF-κB Signaling Pathway after Subarachnoid Hemorrhage in Male Rats. Mediat. Inflamm. 2011, 2011, 848309. [Google Scholar] [CrossRef] [PubMed]
- Hochart, H.; Jenkins, P.V.; Smith, O.P.; White, B. Low-molecular weight and unfractionated heparins induce a downregulation of inflammation: Decreased levels of proinflammatory cytokines and nuclear factor-kappaB in LPS-stimulated human monocytes. Br. J. Haematol. 2006, 133, 62–67. [Google Scholar] [CrossRef]
- Khattar, N.K.; James, R.F. Heparin: The Silver Bullet of Aneurysmal Subarachnoid Hemorrhage? Front. Neurol. 2018, 9, 97. [Google Scholar] [CrossRef]
- Richter, W.F.; Jacobsen, B. Subcutaneous Absorption of Biotherapeutics: Knowns and Unknowns. Drug Metab. Dispos. 2014, 42, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.V.; Drehmer, D.L.; Iacomini, M.; Sassaki, G.L.; Cipriani, T.R. Biological and structural analyses of bovine heparin fractions of intermediate and high molecular weight. Carbohydr. Polym. 2017, 157, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, J.; Snyder, K.A.; Graffeo, C.S.; Khanal, D.R.; Lanzino, G.; Wijdicks, E.F.M.; Rabinstein, A.A. Risk of Ventriculostomy-Associated Hemorrhage in Patients with Aneurysmal Subarachnoid Hemorrhage Treated with Anticoagulant Thromboprophylaxis. Neurocrit. Care 2016, 25, 224–229. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tartara, F.; Montalbetti, A.; Crobeddu, E.; Armocida, D.; Tavazzi, E.; Cardia, A.; Cenzato, M.; Boeris, D.; Garbossa, D.; Cofano, F. Compartmental Cerebrospinal Fluid Events Occurring after Subarachnoid Hemorrhage: An “Heparin Oriented” Systematic Review. Int. J. Mol. Sci. 2023, 24, 7832. https://doi.org/10.3390/ijms24097832
Tartara F, Montalbetti A, Crobeddu E, Armocida D, Tavazzi E, Cardia A, Cenzato M, Boeris D, Garbossa D, Cofano F. Compartmental Cerebrospinal Fluid Events Occurring after Subarachnoid Hemorrhage: An “Heparin Oriented” Systematic Review. International Journal of Molecular Sciences. 2023; 24(9):7832. https://doi.org/10.3390/ijms24097832
Chicago/Turabian StyleTartara, Fulvio, Andrea Montalbetti, Emanuela Crobeddu, Daniele Armocida, Eleonora Tavazzi, Andrea Cardia, Marco Cenzato, Davide Boeris, Diego Garbossa, and Fabio Cofano. 2023. "Compartmental Cerebrospinal Fluid Events Occurring after Subarachnoid Hemorrhage: An “Heparin Oriented” Systematic Review" International Journal of Molecular Sciences 24, no. 9: 7832. https://doi.org/10.3390/ijms24097832