

Oscillation of Autophagy Induction under Cellular Stress and What Lies behind It, a Systems Biology Study
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Time-Delayed Negative Feedback Loop Results in Periodic Activation of Autophagy upon mTOR Inhibition
2.2. Biological Importance of Bistable Switch at Autophagy Induction
2.3. A Direct Negative Feedback Loop of ULK1 Cannot Explain the Periodic Repeat of Autophagy Induction upon Cellular Stress
2.4. The Direct AMPK-ULK1-mTOR Regulatory Triangle Cannot Explain the Periodic Repeat of Autophagy Induction during Rapamycin Treatment
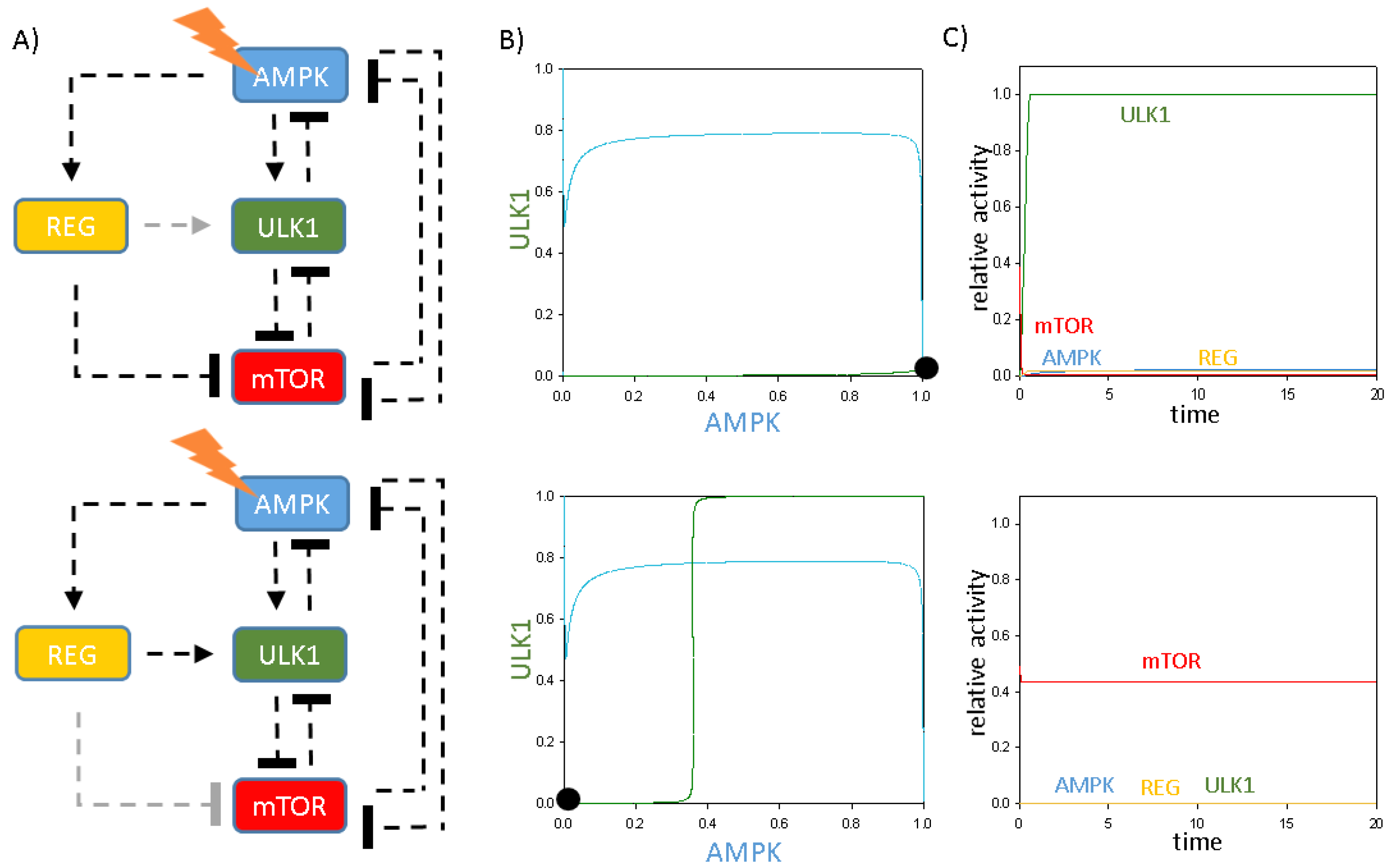
2.5. The Additional Protein Has Direct or Indirect Connections with Its Targets and Regulators
2.6. The Additional Protein Acts like a Key Regulator of Autophagy Induction upon Cellular Stress
3. Discussion
4. Mathematical Models and Methods
4.1. Mathematical Modelling
4.2. Network Analysis Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AMPK | 5′ AMP-activated protein kinase |
| mTORC1 | mammalian target of rapamycin complex 1 |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| FKBP12 | 12-kDa FK506-binding protein |
| ODE | ordinary differential equation |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| TRAF | TNF receptor associated factor |
References
- Eskelinen, E.L. Autophagy: Supporting cellular and organismal homeostasis by self-eating. Int. J. Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int. J. Mol. Sci. 2019, 20, 5543. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.X.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, W.; Nys, K.; Arijs, I.; Cleynen, I.; Noben, M.; De Schepper, S.; Van Assche, G.; Ferrante, M.; Vermeire, S. Biopsy-derived intestinal epithelial cell cultures for pathway-based stratification of patients with inflammatory bowel disease. J. Crohn’s Colitis 2018, 12, 178–187. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and autophagy-related diseases: A review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic targeting of autophagy via HDAC inhibition in tumor cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. Fine-tuning of AMPK–ULK1–mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Hajdú, B.; Holczer, M.; Horváth, G.; Szederkényi, G.; Kapuy, O. Fine-Tuning of mTORC1-ULK1-PP2A Regulatory Triangle Is Crucial for Robust Autophagic Response upon Cellular Stress. Biomolecules 2022, 12, 1587. [Google Scholar] [CrossRef]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Dite, T.A.; Ling, N.X.; Scott, J.W.; Hoque, A.; Galic, S.; Parker, B.L.; Ngoei, K.R.; Langendorf, C.G.; O’Brien, M.T.; Kundu, M.; et al. The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat. Commun. 2017, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef]
- Roach, P.J. AMPK → ULK1 → Autophagy. Mol. Cell. Biol. 2011, 31, 3082–3084. [Google Scholar] [CrossRef]
- Mingione, A.; Ottaviano, E.; Barcella, M.; Merelli, I.; Rosso, L.; Armeni, T.; Cirilli, N.; Ghidoni, R.; Borghi, E.; Signorelli, P. Cystic fibrosis defective response to infection involves autophagy and lipid metabolism. Cells 2020, 9, 1845. [Google Scholar] [CrossRef]
- Nalairndran, G.; Hassan Abdul Razack, A.; Mai, C.W.; Fei-Lei Chung, F.; Chan, K.K.; Hii, L.W.; Lim, W.M.; Chung, I.; Leong, C.O. Phosphoinositide-dependent Kinase-1 (PDPK1) regulates serum/glucocorticoid-regulated Kinase 3 (SGK3) for prostate cancer cell survival. J. Cell. Mol. Med. 2020, 24, 12188–12198. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, Y.; Deng, T.; Gu, J.; Liu, J.; Yang, H.; Xu, Y.; Yan, Y.; Yang, F.; Zhang, H.; et al. PDPK1 regulates autophagosome biogenesis by binding to PIK3C3. Autophagy 2021, 17, 2166–2183. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K. Cdc37 as a co-chaperone to Hsp90. Subcell Biochem. 2015, 78, 103–112. [Google Scholar] [PubMed]
- Zhu, J.; Yan, F.; Tao, J.; Zhu, X.; Liu, J.; Deng, S.; Zhang, X. Cdc37 facilitates cell survival of colorectal carcinoma via activating the CDK 4 signaling pathway. Cancer Sci. 2018, 109, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, L.; Finegold, M.; DeMayo, F.; Schmidt, E.V.; Harper, J.W. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol. Cell. Biol. 2000, 20, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.N.; St Charitoudis, G.; Bechrakis, N.E.; Kozobolis, V.P.; Koukourakis, M.I.; Foerster, M.H.; Sivridis, E.L. Autophagy patterns and prognosis in uveal melanomas. Mod. Pathol. 2011, 24, 1036–1045. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef]
- Bodas, M.; Vij, N. Adapting proteostasis and autophagy for controlling the pathogenesis of cystic fibrosis lung disease. Front. Pharmacol. 2019, 10, 20. [Google Scholar] [CrossRef]
- Guangwei, Z.; Zhibin, C.; Qin, W.; Chunlin, L.; Penghang, L.; Ruofan, H.; Hui, C.; Hoffman, R.M.; Jianxin, Y. TRAF6 regulates the signaling pathway influencing colorectal cancer function through ubiquitination mechanisms. Cancer Sci. 2022, 113, 1393. [Google Scholar] [CrossRef]
- Paul, P.K.; Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy 2011, 7, 555–556. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12: A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995, 13, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Puccetti, P.; Bistoni, F. Interleukin-12 in infectious diseases. Clin. Microbiol. Rev. 1997, 10, 611–636. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kuang, W.; Wu, B.; Xie, C.; Liu, C.; Tu, Z. IL-12 induces autophagy in human breast cancer cells through AMPK and the PI3K/Akt pathway. Mol. Med. Rep. 2017, 16, 4113–4118. [Google Scholar] [CrossRef]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive oxygen species formation in the brain at different oxygen levels: The role of hypoxia inducible factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [PubMed]
- Chang, K.C.; Liu, P.F.; Chang, C.H.; Lin, Y.C.; Chen, Y.J.; Shu, C.W. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci. 2022, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and selective autophagy in cancer development and therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef]
- Novák, B.; Tyson, J.J. Design principles of biochemical oscillators. Nat. Rev. Mol. Cell Biol. 2008, 9, 981–991. [Google Scholar] [CrossRef]
- Paul, S.; Andrew, M.; Owen, O. Baliga Nitin S, Wang Jonathan T, Ramage Daniel, Amin Nada, Schwikowski Benno, Ideker Trey. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar]
- Dixit, V.K.; Rackauckas, C. Globalsensitivity. jl: Performant and parallel global sensitivity analysis with julia. J. Open Source Softw. 2022, 7, 4561. [Google Scholar] [CrossRef]
- Sumner, T. Sensitivity Analysis in Systems Biology Modelling and Its Application to a Multi-Scale Model of Blood Glucose Homeostasis. Ph.D. Thesis, University College London, London, UK, 2010. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajdú, B.; Csabai, L.; Márton, M.; Holczer, M.; Korcsmáros, T.; Kapuy, O. Oscillation of Autophagy Induction under Cellular Stress and What Lies behind It, a Systems Biology Study. Int. J. Mol. Sci. 2023, 24, 7671. https://doi.org/10.3390/ijms24087671
Hajdú B, Csabai L, Márton M, Holczer M, Korcsmáros T, Kapuy O. Oscillation of Autophagy Induction under Cellular Stress and What Lies behind It, a Systems Biology Study. International Journal of Molecular Sciences. 2023; 24(8):7671. https://doi.org/10.3390/ijms24087671
Chicago/Turabian StyleHajdú, Bence, Luca Csabai, Margita Márton, Marianna Holczer, Tamás Korcsmáros, and Orsolya Kapuy. 2023. "Oscillation of Autophagy Induction under Cellular Stress and What Lies behind It, a Systems Biology Study" International Journal of Molecular Sciences 24, no. 8: 7671. https://doi.org/10.3390/ijms24087671