
Coexisting Nodular Sclerosis Hodgkin Lymphoma and Kimura’s Disease: A Case Report and Literature Review
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
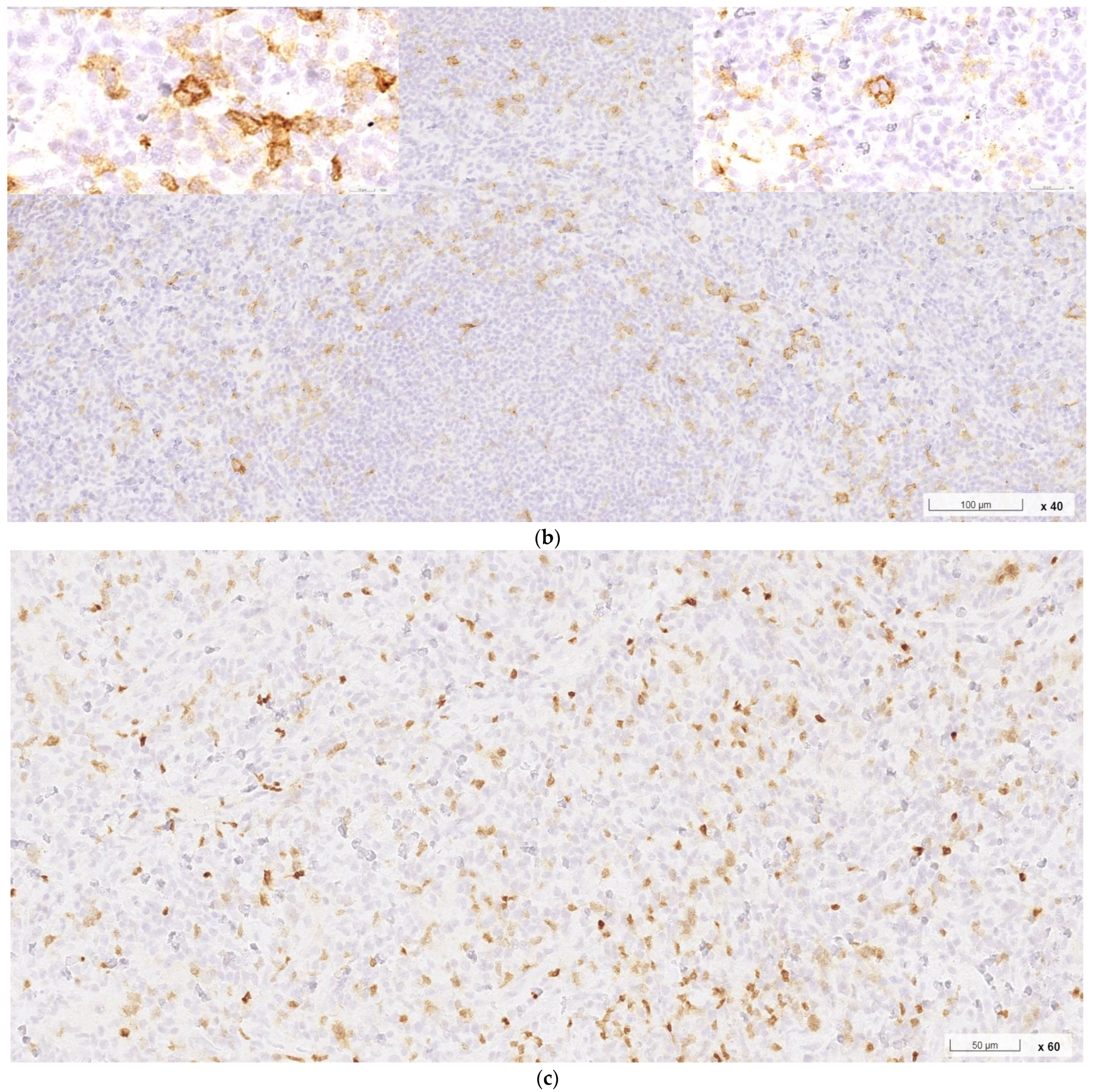
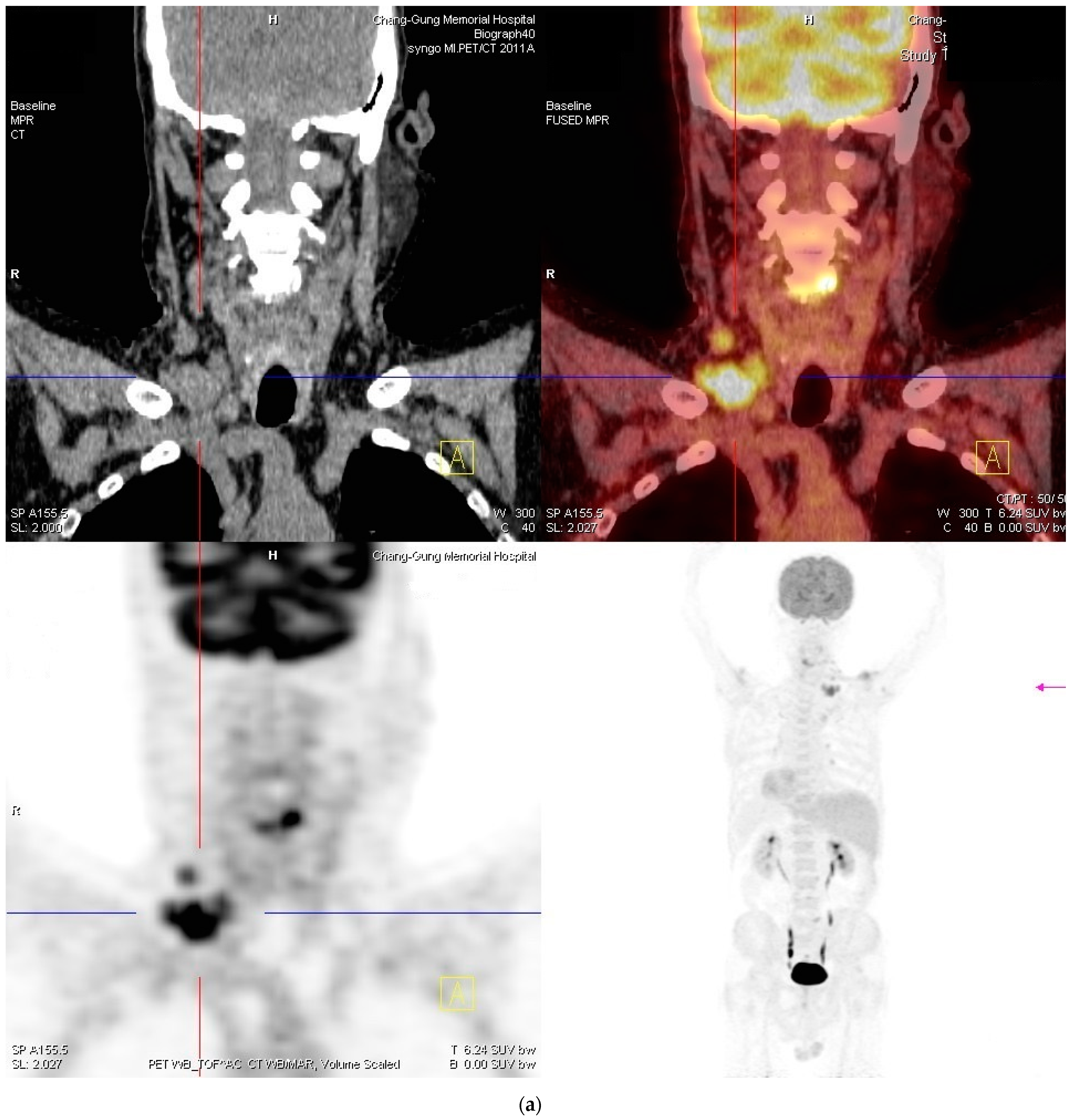
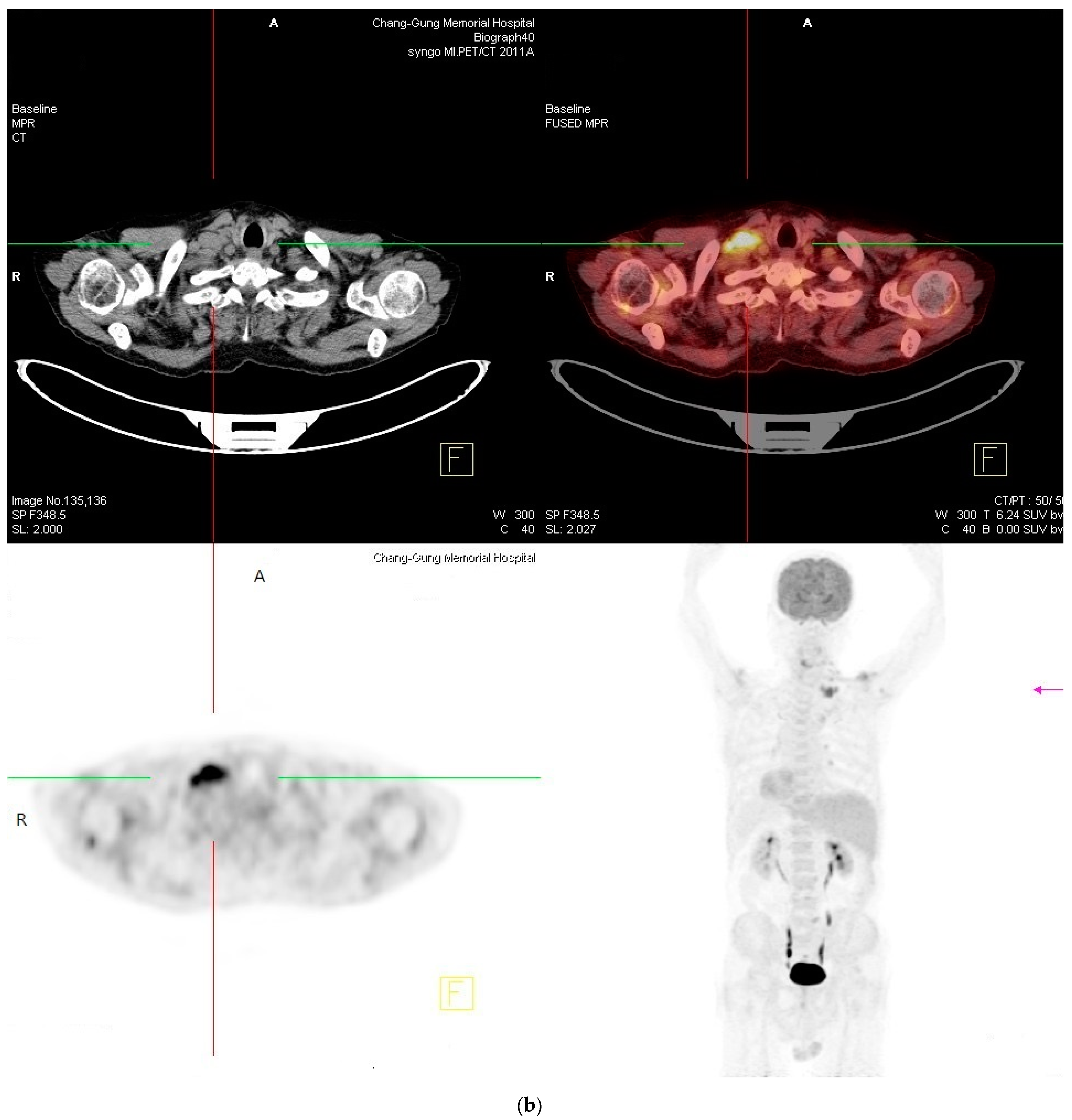
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, C.-C.; Yu, K.-H.; Chan, T.-M. Kimura’s disease: A clinicopathological study of 23 cases. Front. Med. 2022, 9, 1069102. [Google Scholar] [CrossRef] [PubMed]
- Kimm, H.T.; Szeto, C. Eosinophilic hyperplastic lymphogranuloma, comparison with Mikulicz’s disease. Chin. Med. J. 1937, 23, 670–699. [Google Scholar]
- Kimura, T.; Yoshimura, S.; Ishikawa, E. On the unusual granulation combined with hyperplastic changes of lymphatic tissues. Trans. Soc. Pathol. Jpn. 1948, 37, 179–180. [Google Scholar]
- Wang, X.; Ng, C.S.; Yin, W. A comparative study of Kimura’s disease and IgG4-related disease: Similarities, differences and overlapping features. Histopathology 2021, 79, 801–809. [Google Scholar] [CrossRef]
- King, R.L.; Tan, B.; Craig, F.E.; George, T.I.; Horny, H.P.; Kelemen, K.; Orazi, A.; Reichard, K.K.; Rimsza, L.M.; Wang, S.A.; et al. Reactive eosinophil proliferations in tissue and the lymphocytic variant of hypereosin-ophilic syndrome, 2019 Society for Hematopathology/European association for haematopathology workshop report. Am. J. Clin. Pathol. 2020, 155, 211–238. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Yu, G.Y. Clinicopathologic study of parotid involvement in 21 cases of eosinophilic hyperplastic lym-phogranuloma (Kimura’s disease). Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2006, 102, 651–658. [Google Scholar] [CrossRef]
- Hashida, Y.; Higuchi, T.; Nakajima, K.; Ujihara, T.; Murakami, I.; Fujieda, M.; Sano, S.; Daibata, M. Human polyomavirus 6 with the Asian–Japanese Genotype in Cases of Kimura disease and angiolymphoid hyperplasia with eosinophilia. J. Investig. Dermatol. 2020, 140, 1650–1653.e1654. [Google Scholar] [CrossRef]
- Kottler, D.; Barète, S.; Quéreux, G.; Ingen-Housz-Oro, S.; Fraitag, S.; Ortonne, N.; Deschamps, L.; Rybojad, M.; Flageul, B.; Crickx, B.; et al. Retrospective Multicentric Study of 25 Kimura Disease Patients: Emphasis on Therapeutics and Shared Features with Cutaneous IgG4-Related Disease. Dermatology 2015, 231, 367–377. [Google Scholar] [CrossRef]
- Sato, R.; Bandoh, N.; Goto, T.; Ichikawa, H.; Uemura, A.; Suzuki, S.; Yamaguchi, T.; Aimono, E.; Nishihara, H.; Katada, A.; et al. Kimura disease presenting with buccal mass: A case report and literature review. Head Neck Pathol. 2020, 15, 657–662. [Google Scholar] [CrossRef]
- Zhang, G.; Li, X.; Sun, G.; Cao, Y.; Gao, N.; Qi, W. Clinical analysis of Kimura’s disease in 24 cases from China. BMC Surg. 2020, 20, 1. [Google Scholar] [CrossRef]
- Katagiri, K.; Itami, S.; Hatano, Y.; Yamaguchi, T.; Takayasu, S. In vivo expression of IL-4, IL-5, IL-13 and IFN-γ mRNAs in peripheral blood mononuclear cells and effect of cyclosporin A in a patient with Kimura’s disease. Br. J. Dermatol. 1997, 137, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Pawankar, R.; Aoki, M.; Niimi, Y.; Kawana, S. Mast cells and T cells in Kimura’s disease express increased levels of interleukin-4, interleukin-5, eotaxin and RANTES. Clin. Exp. Allergy 2002, 32, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Munemura, R.; Maehara, T.; Murakami, Y.; Koga, R.; Aoyagi, R.; Kaneko, N.; Doi, A.; Perugino, C.A.; Della-Torre, E.; Saeki, T.; et al. Distinct disease-specific Tfh cell populations in 2 different fibrotic diseases: IgG4-related disease and Kimura disease. J. Allergy Clin. Immunol. 2022, 150, 440–455.e17. [Google Scholar] [CrossRef] [PubMed]
- Arul, J.; Senthil, N.; Marappa, L. Unusual and rare case of generalised lymphadenopathy: Kimura’s disease. BMJ Case Rep. 2018, 2018, 225020. [Google Scholar] [CrossRef]
- Gopinathan, A.; Tan, T. Kimura’s disease: Imaging patterns on computed tomography. Clin. Radiol. 2009, 64, 994–999. [Google Scholar] [CrossRef]
- Horikoshi, T.; Motoori, K.; Ueda, T.; Shimofusa, R.; Hanazawa, T.; Okamoto, Y.; Ito, H. Head and neck MRI of Kimura disease. Br. J. Radiol. 2011, 84, 800–804. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Grimm, K.E. Reactive lymphadenopathies that mimic lymphoma: Entities of unknown etiology. Semin. Diagn. Pathol. 2013, 30, 137–145. [Google Scholar] [CrossRef]
- Wang, T.-F.; Liu, S.-H.; Kao, C.-H.K.; Chu, S.-C.; Kao, R.-H.; Li, C.-C. Kimura’s Disease with Generalized LymphadenopathyDemonstrated by Positron Emission Tomography Scan. Intern. Med. 2006, 45, 775–778. [Google Scholar] [CrossRef]
- Yu, M.; Wang, Z.; Zhao, J.; Wu, F. Lymphadenopathy Due to Kimura’s Disease Mimicking Lymphoma on FDG PET/CT. Clin. Nucl. Med. 2019, 44, 299–300. [Google Scholar] [CrossRef]
- Beyazit, Y.; Haznedaroglu, I.C.; Aksu, S.; Kekilli, M.; Uner, A.; Agbaht, K.; Sungur, A.; Koca, E.; Goker, H.; Ozcebe, O.I. Changing clinical manifestations of a T-peripheral lymphoma: From hypereosinophilic syndrome to questionable Kimura’s disease resulting in parotid mass. Leuk. Lymphoma 2006, 47, 357–360. [Google Scholar] [CrossRef]
- Kojima, M.; Yokoo, H.; Yoshida, T.; Jinbo, T.; Nakamura, S. Peripheral T-cell lymphoma resembling Kimura’s disease. Letter to the editor. Apmis 2008, 116, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Fung, A.; Shek, T.W.H.; Liang, R.; Ho, W.K.; Kwong, Y.L. Analysis of Clonality in Kimura’s Disease. Am. J. Surg. Pathol. 2002, 26, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-A.; Ahn, S.-J.; Choi, J.-H.; Sung, K.-J.; Moon, K.-C.; Koh, J.-K.; Shim, Y.-H. Polymerase chain reaction (PCR) for human herpesvirus 8 and heteroduplex PCR for clonality assessment in angiolymphoid hyperplasia with eosinophilia and Kimura’s disease. J. Cutan. Pathol. 2001, 28, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Wei, T.; Yu, G.-Y.; Wu, L.-L.; Peng, X. Comparison of Local Recurrence Rate of Three Treatment Modalities for Kimura Disease. J. Craniofacial Surg. 2016, 27, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Feng, I.-J.; Chen, Y.-T.; Weng, S.-F.; Chan, L.-P.; Lai, C.-S.; Lin, S.-D.; Kuo, Y.-R. Treatment algorithm for Kimura’s disease: A systematic review and meta-analysis of treatment modalities and prognostic predictors. Int. J. Surg. 2022, 100, 106591. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Xu, F.; Zeng, C.; Liu, Z. Clinicopathological features and prognosis of Kimura’s disease with renal involvement in Chinese patients. Clin. Nephrol. 2016, 85, 332–339. [Google Scholar] [CrossRef]
- Ren, S.; Li, X.Y.; Wang, F.; Zhang, P.; Zhang, Y.; Li, G.S.; Wang, L.; Zhong, X. Nephrotic syndrome associated with Kimura’s disease: A case report and literature review. BMC Nephrol. 2018, 19, 316. [Google Scholar] [CrossRef]
- Brice, P.; de Kerviler, E.; Friedberg, J.W. Classical Hodgkin lymphoma. Lancet 2021, 398, 1518–1527. [Google Scholar] [CrossRef]
- Piris, M.A.; Medeiros, L.J.; Chang, K.-C. Hodgkin lymphoma: A review of pathological features and recent advances in pathogenesis. Pathology 2019, 52, 154–165. [Google Scholar] [CrossRef]
- Chuang, S.-S.; Chen, S.-W.; Chang, S.-T.; Kuo, Y.-T. Lymphoma in Taiwan: Review of 1347 neoplasms from a single institution according to the 2016 Revision of the World Health Organization Classification. J. Formos. Med. Assoc. 2017, 116, 620–625. [Google Scholar] [CrossRef]
- Health Promotion Administration Ministry of Health and Welfare Taiwan (n.d.). Cancer Registry Annual Report 2020. Available online: https://www.hpa.gov.tw/File/Attach/16434/File_20339.pdf (accessed on 19 February 2023).
- Akkaya, I.; Oylumlu, E.; Ozel, I.; Uzel, G.; Durmus, L.; Ciraci, C. NLRC4 Inflammasome-Mediated Regulation of Eosinophilic Functions. Immune Netw. 2021, 21, e42. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.; Aslam, I.; Malek, E. Eosinophils upregulate PD-L1 and PD-L2 expression to enhance the immunosuppressive microenvironment in multiple myeloma. Blood 2017, 130, 4417. [Google Scholar]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-C.; Chang, S.-Y.; Teng, W.-C.; Wu, C.-J.; Liu, C.-H.; Huang, S.-W.; Wu, C.-E.; Yu, K.-H.; Chan, T.-M. Coexisting Nodular Sclerosis Hodgkin Lymphoma and Kimura’s Disease: A Case Report and Literature Review. Int. J. Mol. Sci. 2023, 24, 7666. https://doi.org/10.3390/ijms24087666
Lee C-C, Chang S-Y, Teng W-C, Wu C-J, Liu C-H, Huang S-W, Wu C-E, Yu K-H, Chan T-M. Coexisting Nodular Sclerosis Hodgkin Lymphoma and Kimura’s Disease: A Case Report and Literature Review. International Journal of Molecular Sciences. 2023; 24(8):7666. https://doi.org/10.3390/ijms24087666
Chicago/Turabian StyleLee, Chih-Chun, Sing-Ya Chang, Wen-Chieh Teng, Chih-Ju Wu, Chi-Hung Liu, Szu-Wei Huang, Chiao-En Wu, Kuang-Hui Yu, and Tien-Ming Chan. 2023. "Coexisting Nodular Sclerosis Hodgkin Lymphoma and Kimura’s Disease: A Case Report and Literature Review" International Journal of Molecular Sciences 24, no. 8: 7666. https://doi.org/10.3390/ijms24087666