
Vascular Stem Cells and the Role of B-Raf Kinase in Survival, Proliferation, and Apoptosis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Fetal Arteries Grow in Serum-Free Media
2.2. De Novo Vessel Formation from VSCs
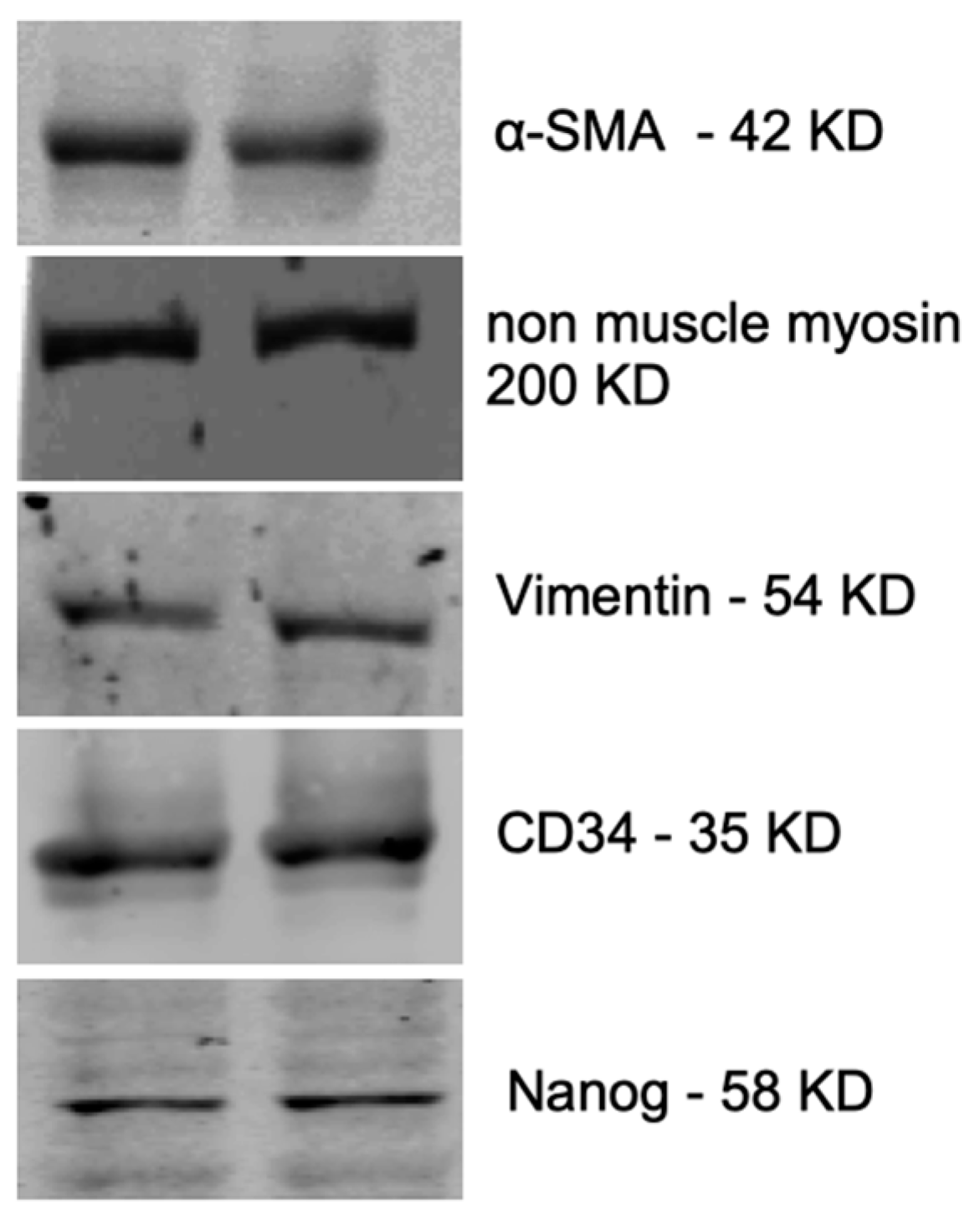
2.3. Characterization of Fetal Cells Growing from Carotid Artery Segments
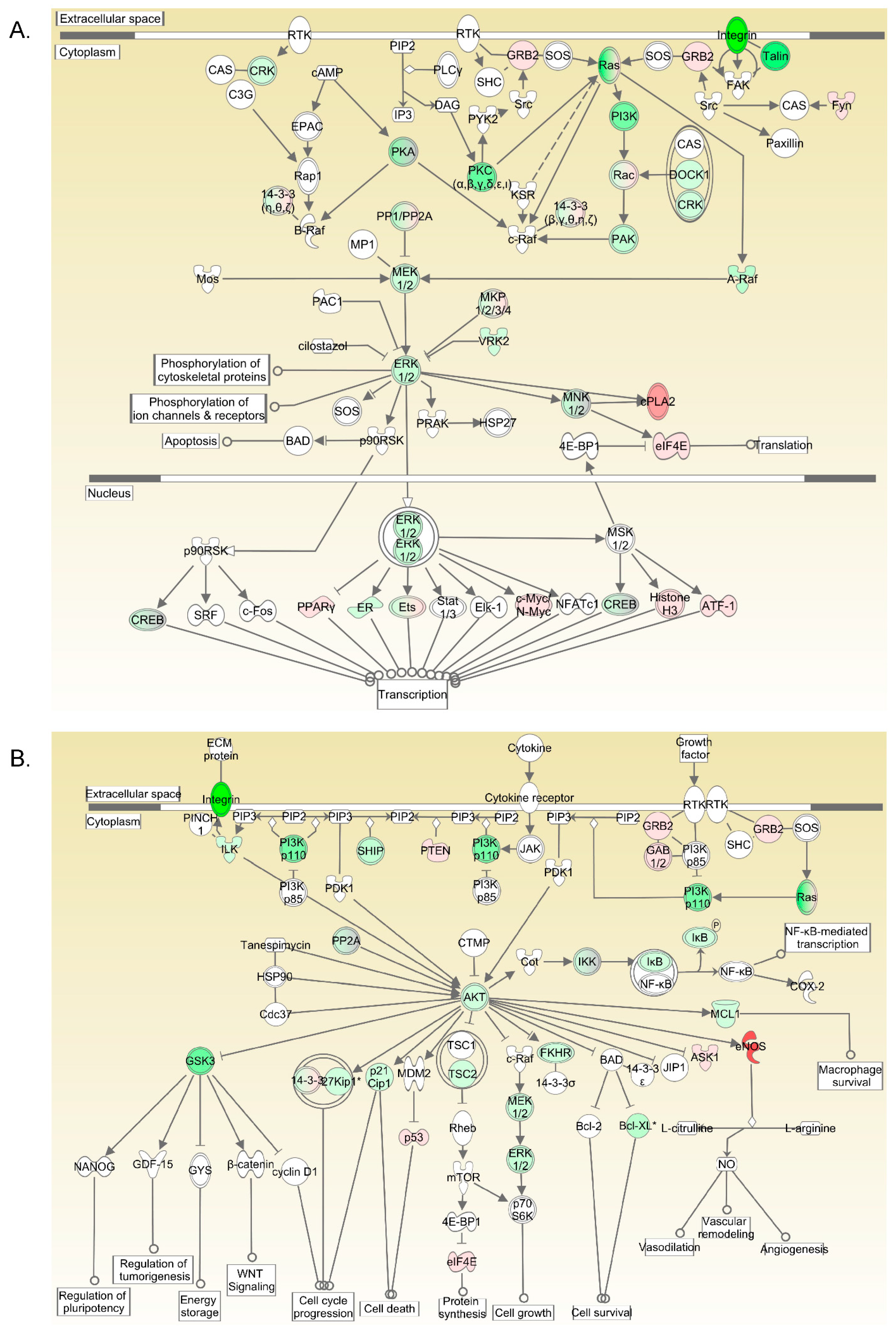
2.4. Kinase Pathways Are Upregulated in Fetus Arterial Segments
2.5. BRAF Inhibition Results in Vacuolar Degeneration of Cells
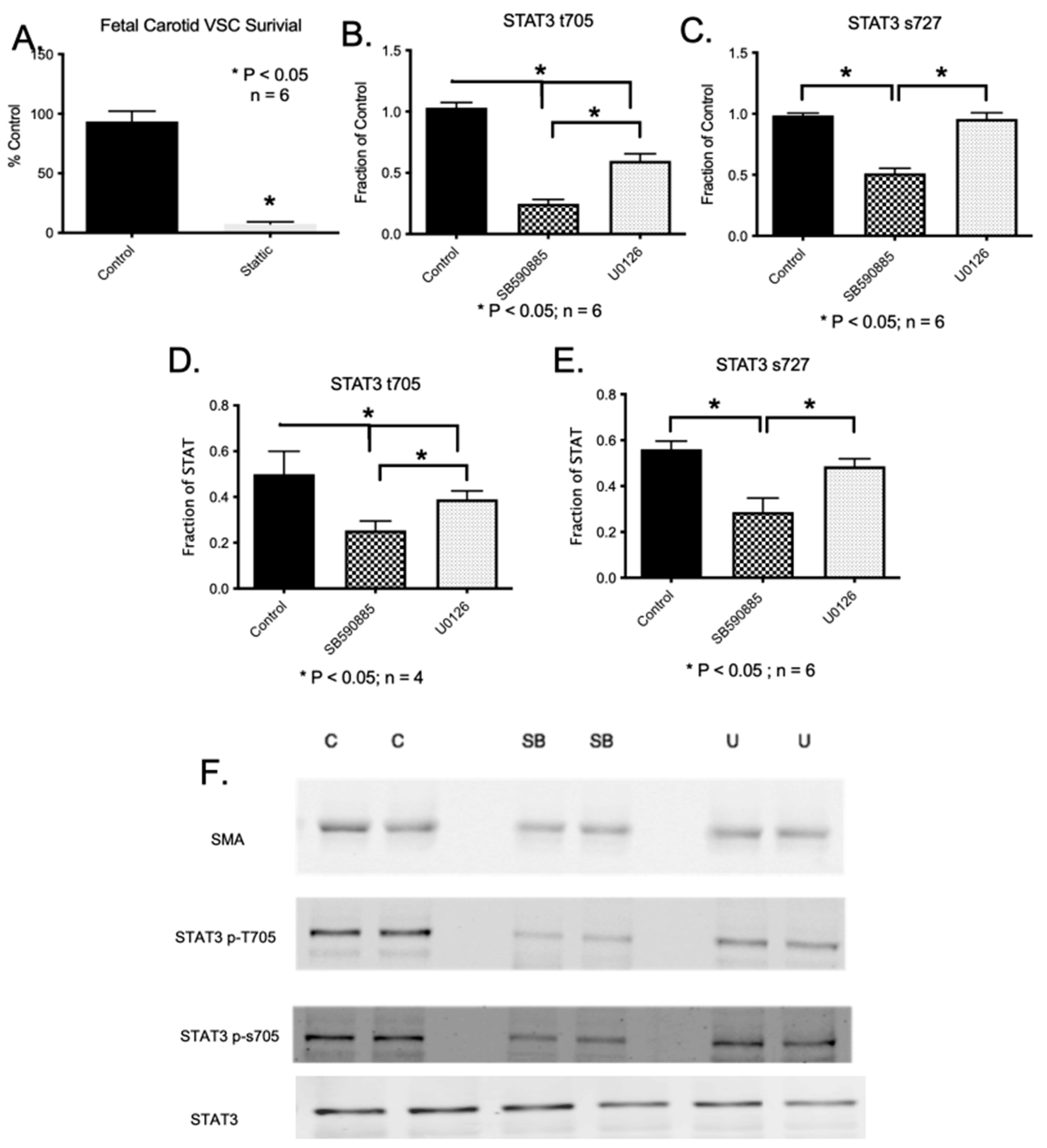
2.6. BRAF Inhibition Results in Increased Cell Apoptosis
2.7. B-Raf Inhibition Leads to Vacuolar Cell Death of Fetal VSC
2.8. B-Raf Inhibition Leads to Cell Cycle Arrest in G1/Go Phase
2.9. Downstream Pathways of B-Raf Mediated VSC Cell Viability and Survival
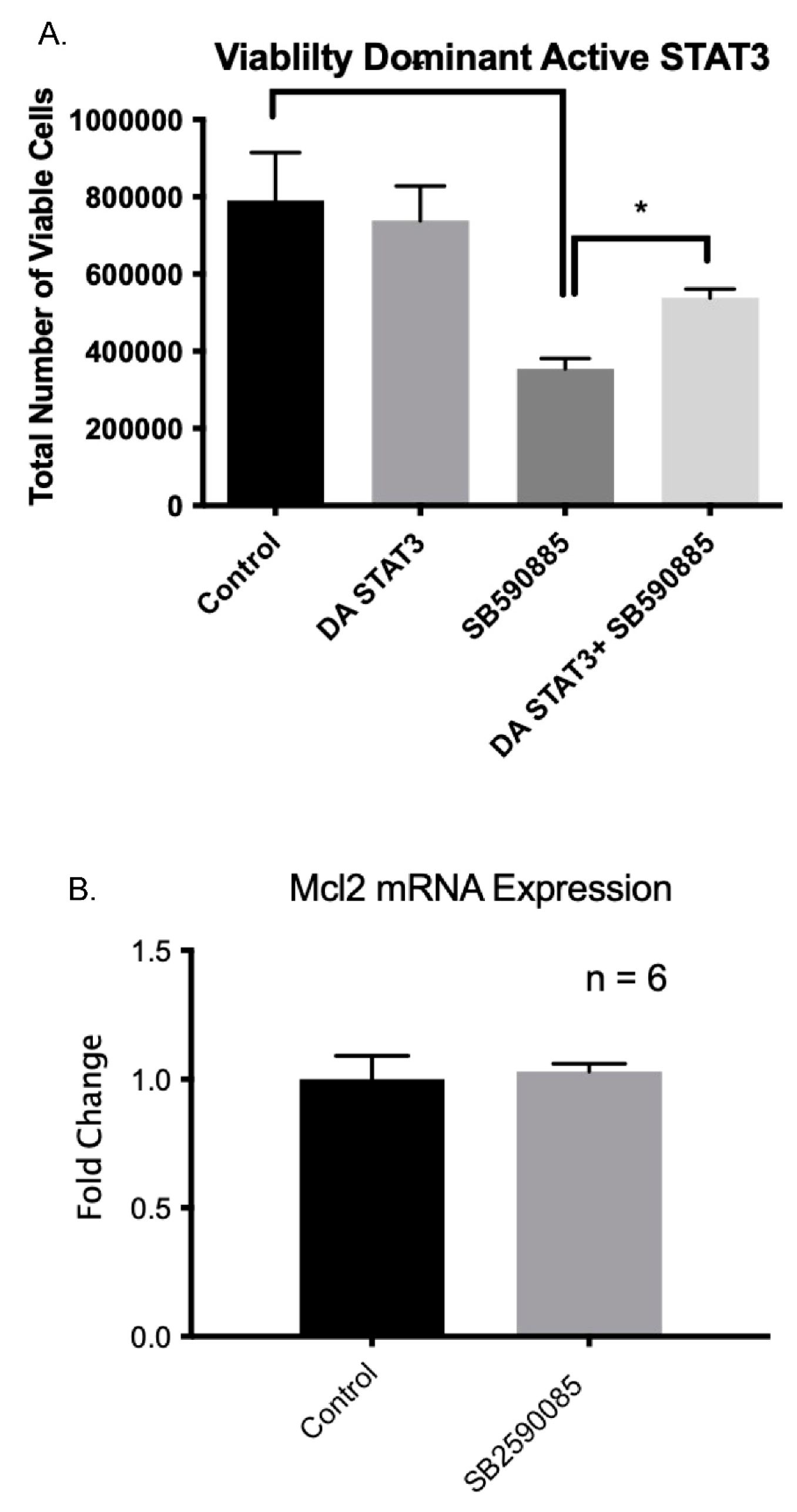
2.10. Constitutive Active STAT3 Prevents B-Raf-Induced Apoptosis
2.11. B-Raf Inhibition Leads to the Inhibition of Bcl2 Gene Expression
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Tissues
4.2. Carotid Artery VSC Growth and Isolation
4.3. Immunoblot Procedures
4.4. Epifluorescence Imaging Studies
4.5. Cell Viability and Proliferation Assay
4.6. Apoptosis Assay
4.7. Cell-Cycle Assay
4.8. Real-Time PCR
4.9. Transcriptomic Analysis
4.10. Transfection of Constitutive Active STAT3
4.11. Endothelial Cell Isolation and In Vitro Tube Formation Assay
4.12. Statistical Analysis
4.13. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Issa Bhaloo, S.; Chen, T.; Zhou, B.; Xu, Q. Role of Resident Stem Cells in Vessel Formation and Arteriosclerosis. Circ. Res. 2018, 122, 1608–1624. [Google Scholar] [CrossRef] [PubMed]
- Lahteenvuo, J.; Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 2012, 110, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Bonham, C.A.; Kuehlmann, B.; Gurtner, G.C. Impaired Neovascularization in Aging. Adv. Wound. Care 2020, 9, 111–126. [Google Scholar] [CrossRef]
- Lin, C.S.; Lue, T.F. Defining vascular stem cells. Stem. Cells Dev. 2013, 22, 1018–1026. [Google Scholar] [CrossRef]
- Tao, J.; Cao, X.; Yu, B.; Qu, A. Vascular Stem/Progenitor Cells in Vessel Injury and Repair. Front. Cardiovasc. Med. 2022, 9, 845070. [Google Scholar] [CrossRef]
- Barachini, S.; Ghelardoni, S.; Madonna, R. Vascular Progenitor Cells: From Cancer to Tissue Repair. J. Clin. Med. 2023, 12, 2399. [Google Scholar] [CrossRef]
- Han, S.; Sun, H.M.; Hwang, K.C.; Kim, S.W. Adipose-Derived Stromal Vascular Fraction Cells: Update on Clinical Utility and Efficacy. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 145–152. [Google Scholar] [CrossRef]
- van Beijnum, J.R.; Rousch, M.; Castermans, K.; van der Linden, E.; Griffioen, A.W. Isolation of endothelial cells from fresh tissues. Nat. Protoc. 2008, 3, 1085–1091. [Google Scholar] [CrossRef]
- Goyal, D.; Goyal, R. Angiogenic Transformation in Human Brain Micro Endothelial Cells: Whole Genome DNA Methylation and Transcriptomic Analysis. Front. Physiol. 2019, 10, 1502. [Google Scholar] [CrossRef]
- Mata-Greenwood, E.; Goyal, D.; Goyal, R. Comparative and Experimental Studies on the Genes Altered by Chronic Hypoxia in Human Brain Microendothelial Cells. Front. Physiol. 2017, 8, 365. [Google Scholar] [CrossRef]
- Takle, A.K.; Bamford, M.J.; Davies, S.; Davis, R.P.; Dean, D.K.; Gaiba, A.; Irving, E.A.; King, F.D.; Naylor, A.; Parr, C.A.; et al. The identification of potent, selective and CNS penetrant furan-based inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2008, 18, 4373–4376. [Google Scholar] [CrossRef]
- Takle, A.K.; Brown, M.J.B.; Davies, S.; Dean, D.K.; Francis, G.; Gaiba, A.; Hird, A.W.; King, F.D.; Lovell, P.J.; Naylor, A.; et al. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2006, 16, 378–381. [Google Scholar] [CrossRef]
- Duncia, J.V.; Santella, I.; Joseph, B.; Higley, C.A.; Pitts, W.J.; Wityak, J.; Frietze, W.E.; Rankin, F.W.; Sun, J.-H.; Earl, R.A.; et al. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett. 1998, 8, 2839–2844. [Google Scholar] [CrossRef]
- Ishizaki, T.; Uehata, M.; Tamechika, I.; Keel, J.; Nonomura, K.; Maekawa, M.; Narumiya, S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 2000, 57, 976–983. [Google Scholar]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 1996, 271, 17920–17926. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.J. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 1998, 392, 622–626. [Google Scholar] [CrossRef]
- Becker, T.; Boyd, S.; Mijatov, B.; Gowrishankar, K.; Snoyman, S.; Pupo, G.; Scolyer, R.; Mann, G.; Kefford, R.; Zhang, X.; et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene 2014, 33, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Oka, M.; Iwasaki, T.; Fukami, Y.; Nishigori, C. Role and Regulation of STAT3 Phosphorylation at Ser727 in Melanocytes and Melanoma Cells. J. Investig. Dermatol. 2012, 132, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.; James, E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef]
- Wang, J.-L.; Liu, D.; Zhang, Z.-J.; Shan, S.; Han, X.; Srinivasula, S.M.; Croce, C.M.; Alnemri, E.S.; Huang, Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7124. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ray, R.M.; Johnson, L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005, 392, 335–344. [Google Scholar] [CrossRef]
- Goyal, R.; Mittal, A.; Chu, N.; Shi, L.; Zhang, L.; Longo, L.D. Maturation and the role of PKC-mediated contractility in ovine cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2242–H2252. [Google Scholar] [CrossRef]
- Schirrmacher, E.; Beck, C.; Brueckner, B.; Schmitges, F.; Siedlecki, P.; Bartenstein, P.; Lyko, F.; Schirrmacher, R. Synthesis and in Vitro Evaluation of Biotinylated RG108:¬† A High Affinity Compound for Studying Binding Interactions with Human DNA Methyltransferases. Bioconjug. Chem. 2006, 17, 261–266. [Google Scholar] [CrossRef]
- Jaworska, J.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The potential neuroprotective role of a histone deacetylase inhibitor, sodium butyrate, after neonatal hypoxia-ischemia. J. Neuroinflamm. 2017, 14, 34. [Google Scholar] [CrossRef]
- Goyal, R.; Henderson, D.A.; Chu, N.; Longo, L.D. Ovine Middle Cerebral Artery Characterization and Quantification of Ultrastructure and Other Features: Changes with Development. AJP Regul. Integr. Comp. Physiol. 2012, 302, R433–R445. [Google Scholar] [CrossRef]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef]
- Akula, S.; Ford, P.; Whitman, A.; Hamden, K.; Bryan, B.; Cook, P.; McCubrey, J. B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood 2005, 105, 4516–4522. [Google Scholar] [CrossRef]
- Alavi, A.; Hood, J.; Frausto, R.; Stupack, D.; Cheresh, D. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 2003, 301, 94–96. [Google Scholar] [CrossRef]
- Hagemann, C.; Gloger, J.; Anacker, J.; Said, H.; Gerngras, S.; Kuhnel, S.; Meyer, C.; Rapp, U.; Kammerer, U.; Vordermark, D.; et al. RAF expression in human astrocytic tumors. Int. J. Mol. Med. 2009, 23, 17–31. [Google Scholar] [CrossRef]
- Galabova-Kovacs, G.; Matzen, D.; Piazzolla, D.; Meissl, K.; Plyushch, T.; Chen, A.P.; Silva, A.; Baccarini, M. Essential role of B-Raf in ERK activation during extraembryonic development. Proc. Natl. Acad. Sci. USA 2006, 103, 1325–1330. [Google Scholar] [CrossRef]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef]
- Goyal, D.; Goyal, R. Developmental Maturation and Alpha-1 Adrenergic Receptors-Mediated Gene Expression Changes in Ovine Middle Cerebral Arteries. Sci. Rep. 2018, 8, 1772. [Google Scholar] [CrossRef]
- Goyal, R.; Goyal, D.; Chu, N.; van Wickle, J.; Longo, L. Cerebral Artery Alpha-1 AR Subtypes: High Altitude Long-Term Acclimatization Responses. PLoS ONE 2014, 9, e112784. [Google Scholar] [CrossRef]
- Goyal, R.; Longo, L.D. Gene Expression in Sheep Carotid Arteries: Major Changes with Maturational Development. Pediatr. Res. 2012, 72, 137–146. [Google Scholar] [CrossRef]
- Goyal, R.; Galffy, A.; Field, S.A.; Gheorghe, C.P.; Mittal, A.; Longo, L.D. Maternal protein deprivation: Changes in systemic renin-angiotensin system of the mouse fetus. Reprod. Sci. 2009, 16, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids. Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Bunnell, B.A.; Flaat, M.; Gagliardi, C.; Patel, B.; Ripoll, C. Adipose-derived stem cells: Isolation, expansion and differentiation. Methods 2008, 45, 115–120. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goyal, D.; Limesand, S.W.; Goyal, R. Vascular Stem Cells and the Role of B-Raf Kinase in Survival, Proliferation, and Apoptosis. Int. J. Mol. Sci. 2023, 24, 7483. https://doi.org/10.3390/ijms24087483
Goyal D, Limesand SW, Goyal R. Vascular Stem Cells and the Role of B-Raf Kinase in Survival, Proliferation, and Apoptosis. International Journal of Molecular Sciences. 2023; 24(8):7483. https://doi.org/10.3390/ijms24087483
Chicago/Turabian StyleGoyal, Dipali, Sean W. Limesand, and Ravi Goyal. 2023. "Vascular Stem Cells and the Role of B-Raf Kinase in Survival, Proliferation, and Apoptosis" International Journal of Molecular Sciences 24, no. 8: 7483. https://doi.org/10.3390/ijms24087483