
Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets
 , , , and
, , , and
Abstract
:1. Introduction
2. Cellular Invasion Mechanisms
3. Tumor Microenvironment
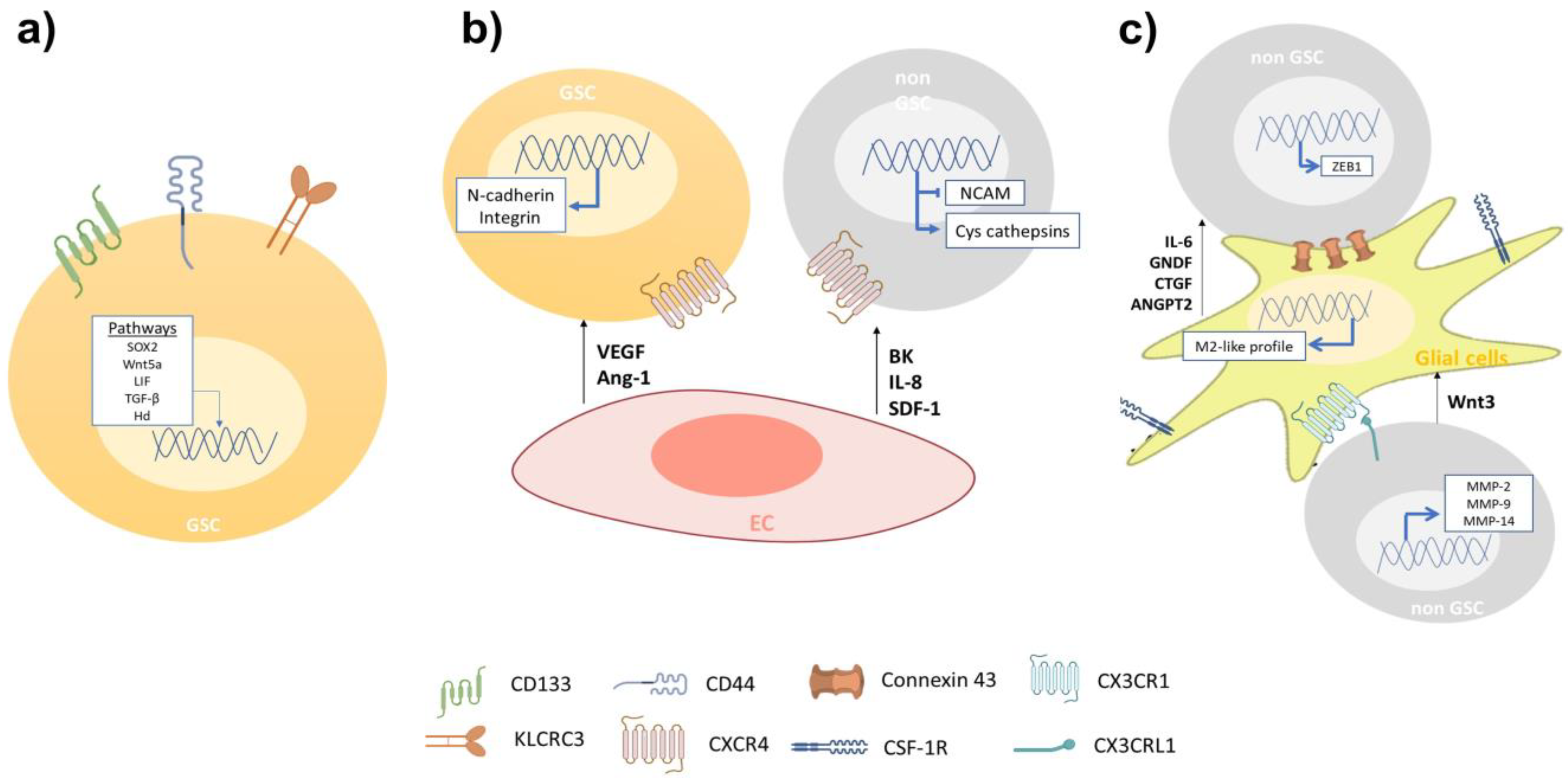
3.1. Cellular Components Involved in GBM Cell Invasion
3.1.1. Glioblastoma Stem-Like Cells
3.1.2. Endothelial Cells
3.1.3. Glial Cells

{kind=link}
{kind=link}
{kind=link}
| Source | Molecule | Mechanism | Reference |
|---|---|---|---|
| GSCs | CD133 |
| [15,16] |
| CD44 |
| [17,18] | |
| KLRC3 |
| [19] | |
| SOX2 |
| [21] | |
| Wnt5a |
| [25,26,27] | |
| LIF |
| [28,29] | |
| TGF-β |
| [30] | |
| Hh |
| [31] | |
| Endothelial cells | VEGF |
| [34,35] |
| Ang-1 |
| [36] | |
| BK |
| [37] | |
| CXCR4 |
| [38] | |
| IL-8 |
| [39] | |
| SDF-1 |
| [40] | |
| Glial cells | IL-6 |
| [43] |
| GNDF |
| [44] | |
| CTGF |
| [45] | |
| Cx43 |
| [48,49] | |
| Ang-2 |
| [51] | |
| CX3CL1/CX3CR1 |
| [63,64] | |
| CSF-1R |
| [65] | |
| Wnt3a |
| [67,68] |
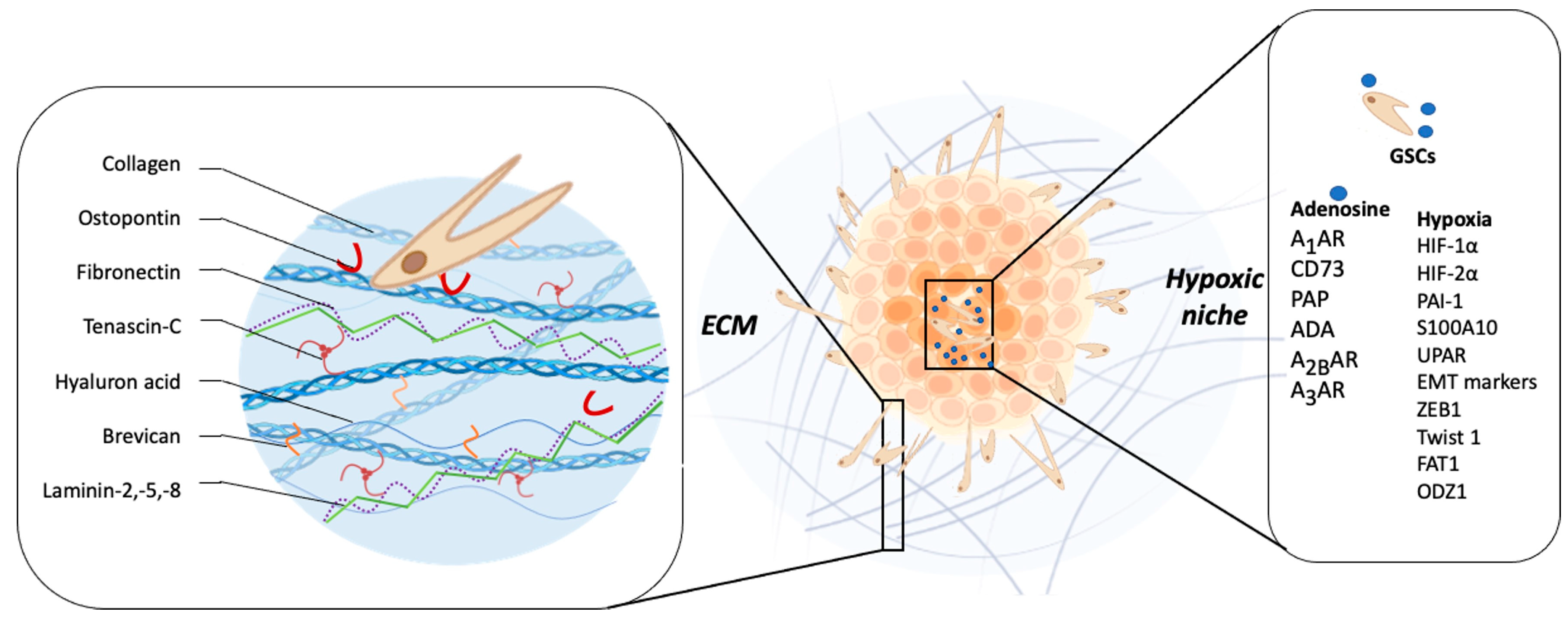
3.2. Non-Cellular Components
3.2.1. Extracellular Matrix
3.2.2. Hypoxia
3.2.3. Adenosine
3.2.4. Senescence and Glioblastoma
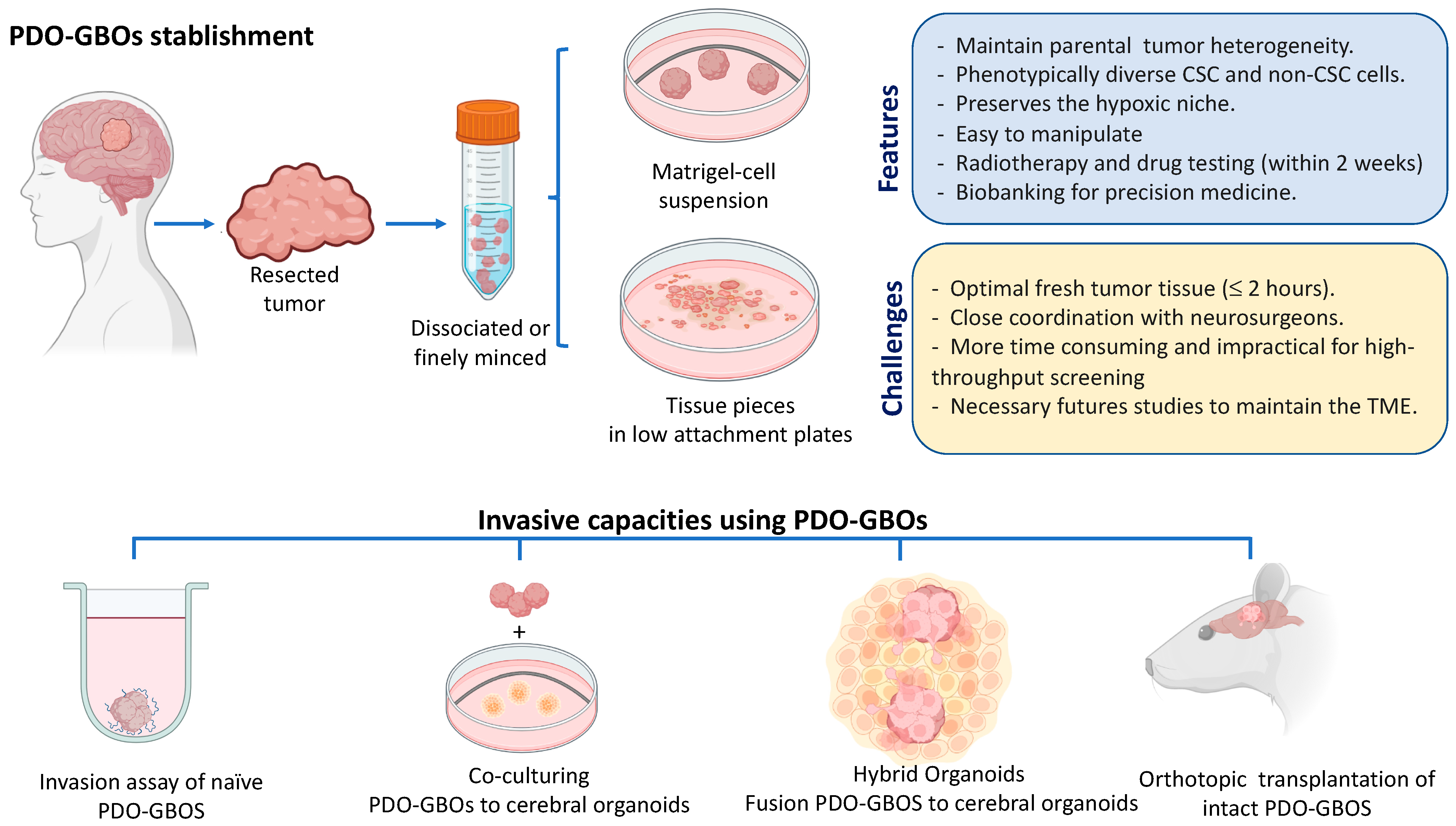
4. Patient-Derived Glioblastoma Organoids for Modeling of Tumor Microenvironment and Invasiveness
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappatz, J.; Norden, A.D.; Wen, P.Y. Therapeutic strategies for inhibiting invasion in glioblastoma. Expert Rev. Neurother. 2009, 9, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from in Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms Regulating Glioma Invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Cayre, M.; Canoll, P.; Goldman, J.E. Cell migration in the normal and pathological postnatal mammalian brain. Prog. Neurobiol. 2009, 88, 41–63. [Google Scholar] [CrossRef] [Green Version]
- Mbeunkui, F.; Johann, D.J., Jr. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Jingzhou, Z.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 15, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso-Sacido, A.; Moliterno, J.A.; Kratovac, S.; Kapoor, G.S.; O’Rourke, D.M.; Holland, E.C.; García-Verdugo, J.M.; Roy, N.S.; Boockvar, J.A. Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. Neurooncol. 2010, 97, 323–337. [Google Scholar] [CrossRef]
- Yu, S.; Yang, X.; Zhang, B.; Ming, H.; Chen, C.; Ren, B.; Liu, Z.; Liu, B. Enhanced invasion in vitro and the distribution patterns in vivo of CD133+ glioma stem cells. Chin. Med. J. 2011, 124, 2599–2604. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 23, 5387041. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Matsumoto, S.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; et al. Hypoxia-regulated expression of CD44 and osteopontin can change the phenotype of glioma stem-like cells from highly invasive to less invasive/proliferative tumors in glioblastoma. Transl. Oncol. 2021, 4, 101137. [Google Scholar] [CrossRef]
- Cheray, M.; Bessette, B.; Lacroix, A.; Mélin, C.; Jawhari, S.; Pinet, S.; Deluche, E.; Clavère, P.; Durand, K.; Sanchez-Prieto, R.; et al. KLRC3, a Natural Killer receptor gene, is a key factor involved in glioblastoma tumourigenesis and aggressiveness. J. Cell. Mol. Med. 2017, 21, 244–253. [Google Scholar] [CrossRef]
- Ortensi, B.; Setti, M.; Osti, D.; Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell. Res. Ther. 2013, 28, 18. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE 2011, 6, e26740. [Google Scholar] [CrossRef] [PubMed]
- Tamase, A.; Muraguchi, T.; Naka, K.; Tanaka, S.; Kinoshita, M.; Hoshii, T.; Ohmura, M.; Shugo, H.; Ooshio, T.; Nakada, M.; et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc. Natl. Acad. Sci. USA 2009, 106, 17163–17168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75, 385–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnés, M.; Casas Tintó, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Kamino, M.; Kishida, M.; Kibe, T.; Ikoma, K.; Iijima, M.; Hirano, H.; Tokudome, M.; Chen, L.; Koriyama, C.; Yamada, K.; et al. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Sci. 2011, 102, 540–548. [Google Scholar] [CrossRef]
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.S.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009, 15, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef]
- Zhu, H.; Lo, H. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genom. 2010, 11, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Maiti, S.; Mondal, S.; Satyavarapu, E.; Mandal, C. mTORC2 regulates hedgehog pathway activity by promoting stability to Gli2 protein and its nuclear translocation. Cell Death Dis. 2017, 8, e2926. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Mao, S.; Li, H.; Zheng, M.; Yi, L.; Lin, J.M.; Lin, Z.X. The pathological structure of the perivascular niche in different microvascular patterns of glioblastoma. PloS ONE 2017, 12, e0182183. [Google Scholar] [CrossRef] [Green Version]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [Green Version]
- Gerigk, M.; Bulstrode, H.; Shi, H.H.; Tönisen, F.; Cerutti, C.; Morrison, G.; Rowitch, D.; Huang, Y.Y.S. On-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture. Lab Chip 2021, 21, 2343–2358. [Google Scholar] [CrossRef]
- Liu, D.; Martin, V.; Fueyo, J.; Lee, O.H.; Xu, J.; Cortes-Santiago, N.; Alonso, M.M.; Aldape, K.; Colman, H.; Gomez-Manzano, C. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget 2010, 1, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83837. [Google Scholar] [CrossRef] [Green Version]
- Coy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.R.; Zipfel, W.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef] [Green Version]
- Kenig, S.; Alonso, M.B.; Mueller, M.M.; Lah, T.T. Glioblastoma and endothelial cells crosstalk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010, 1, 53–61. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 2016, 7, 62425–62438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 2015, 137, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.A.; Woolard, K.; Son, M.J.; Li, A.; Lee, J.; Ene, C.; Mantey, S.A.; Maric, D.; Song, H.; Belova, G.; et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion, n. J. Natl. Cancer Inst. 2011, 103, 1162–1178. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhou, X.W.M.; Wang, X.; Yang, Y.; Luo, J.W.; Liu, Y.H.; Mao, Q. Complex role of connexin 43 in astrocytic tumors and possible promotion of glioma-associated epileptic discharge (Review). Mol. Med. Rep. 2017, 16, 7890–7900. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.; Aftab, Q.; Bechberger, J.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Bercury, K.K.; Macklin, W.B. Dynamics and mechanisms of CNS myelination. Dev. Cell. 2015, 32, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Hide, T.; Shibahara, I.; Kumabe, T. Novel concept of the border niche: Glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol. 2019, 36, 63–73. [Google Scholar] [CrossRef]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef]
- Kawashima, T.; Yashiro, M.; Kasashima, H.; Terakawa, Y.; Uda, T.; Nakajo, K.; Umaba, R.; Tanoue, Y.; Tamrakar, S.; Ohata, K. Oligodendrocytes Up-regulate the Invasive Activity of Glioblastoma Cells via the Angiopoietin-2 Signaling Pathway. Anticancer Res. 2019, 39, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Prionisti, I.; Bühler, L.H.; Walker, P.R.; Jolivet, R.B. Harnessing Microglia and Macrophages for the Treatment of Glioblastoma. Front. Pharmacol. 2019, 10, 506. [Google Scholar] [CrossRef]
- Strepkos, D.; Markouli, M.; Klonou, A.; Piperi, C.; Papavassiliou, A.G. Insights in the immunobiology of glioblastoma. J. Mol. Med. 2020, 98, 1–10. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Invest. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Hotchkiss, K.M.; Patel, K.K.; Wilkinson, D.S.; Mohan, A.A.; Cook, S.L.; Sampson, J.H. Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers 2021, 13, 5367. [Google Scholar] [CrossRef]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.; Hu, M.; Li, P.; Ma, J.; Xie, L.; Teng, F.M.; Zhu, Y.; Fan, B.; Mu, D.; Yu, J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget 2017, 30, 49702–49712. [Google Scholar] [CrossRef] [Green Version]
- Burster, T.; Gärtner, F.; Bulach, C.M.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, A.; Collins, A.; Khoshbouei, H.; Streit, W.J. Microglia and Other Myeloid Cells in Central Nervous System Health and Disease. J. Pharmacol. Exp. Ther. 2020, 375, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held-Feindt, J.; Hattermann, K.; Müerköster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell. Res. 2010, 316, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Matias, D.; Dubois, L.G.; Pontes, B.; Rosário, L.; Ferrer, V.P.; Balça-Silva, J.; Fonseca, A.C.C.; Macharia, L.W.; Romão, L.E.; Spohr, T.C.L.S.; et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/β-Catenin Signaling. Mol. Neurobiol. 2019, 56, 1517–1530. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [Green Version]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 19, 1151. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef]
- Cui, X.; Wang, Q.; Zhou, J.; Wang, Y.; Xu, C.; Tong, F.; Wang, H.; Kang, C. Single-Cell Transcriptomics of Glioblastoma Reveals a Unique Tumor Microenvironment and Potential Immunotherapeutic Target Against Tumor-Associated Macrophage. Front. Oncol. 2021, 11, 710695. [Google Scholar] [CrossRef]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Novak, U.; Kaye, A.H. Extracellular matrix, and the brain: Components and function. J. Clin. Neurosci. 2000, 7, 280–290. [Google Scholar] [CrossRef]
- Ferrer, V.P.; Moura Neto, V.; Mentlein, R. Glioma infiltration and extracellular matrix: Key players and modulators. Glia 2018, 66, 1542–1565. [Google Scholar] [CrossRef]
- Peng, W.J.; Yan, J.W.; Wan, Y.N.; Wang, B.X.; Tao, J.H.; Yang, G.J.; Pan, H.F.; Wang, J. Matrix metalloproteinases: A review of their structure and role in systemic sclerosis. J. Clin. Immunol. 2012, 32, 1409–1414. [Google Scholar] [CrossRef]
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67–79. [Google Scholar] [CrossRef]
- Wiranowska, M.; Ladd, S.; Moscinski, L.C.; Hill, B.; Haller, E.; Mikecz, K.; Plaas, A. Modulation of hyaluronan production by CD44 positive glioma cells. Int. J. Cancer 2010, 127, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annabi, B.; Bouzeghrane, M.; Moumdjian, R.; Moghrabi, A.; Béliveau, R. Probing the infiltrating character of brain tumors: Inhibition of RhoA/ROK-mediated CD44 cell surface shedding from glioma cells by the green tea catechin EGCg. J. Neurochem. 2005, 94, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeckm, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adh. Migr. 2015, 9, 48–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Behrem, S.; Zarković, K.; Eskinja, N.; Jonjić, N. Distribution pattern of tenascin-C in glioblastoma: Correlation with angiogenesis and tumor cell proliferation. Pathol. Oncol. Res. 2005, 11, 229–235. [Google Scholar] [CrossRef]
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro Oncol. 2015, 17, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Ljubimova, J.Y.; Lakhter, A.J.; Loksh, A.; Yong, W.H.; Riedinger, M.S.; Miner, J.H.; Sorokin, L.M.; Ljubimov, A.V.; Black, K.L. Overexpression of alpha4 chain-containing laminins in human glial tumors identified by gene microarray analysis. Cancer Res. 2001, 15, 5601–5610. [Google Scholar]
- Miner, J.H.; Yurchenco, P.D. Laminin functions in tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 2004, 20, 255–284. [Google Scholar] [CrossRef] [Green Version]
- Ljubimova, J.Y.; Fujita, M.; Khazenzon, N.M.; Ljubimov, A.V.; Black, K.L. Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front. Biosci. 2006, 11, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Ljubimova, J.Y.; Fugita, M.; Khazenzon, N.M.; Das, A.; Pikul, B.B.; Newman, D.; Sekiguchi, K.; Sorokin, L.M.; Sasaki, T.; Black, K.L. Association between laminin-8 and glial tumor grade, recurrence, and patient survival. Cancer 2004, 101, 604–612. [Google Scholar] [CrossRef]
- Guo, P.; Imanishi, Y.; Cackowski, F.C.; Jarzynka, M.J.; Tao, H.Q.; Nishikawa, R.; Hirose, T.; Hu, B.; Cheng, S.Y. Up-regulation of angiopoietin-2, matrix metalloprotease-2, membrane type 1 metalloprotease, and laminin 5 gamma 2 correlates with the invasiveness of human glioma. Am. J. Pathol. 2005, 166, 877–890. [Google Scholar] [CrossRef]
- Zhang, H.; Kelly, G.; Zerillo, C.; Jaworski, D.M.; Hockfield, S. Expression of a cleaved brain-specific extracellular matrix protein mediates glioma cell invasion In vivo. J. Neurosci. 1998, 18, 2370–2376. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, C.A.; Bi, W.L.; Viapiano, M.S.; Matthews, R.T. Brevican knockdown reduces late-stage glioma tumor aggressiveness. J. Neurooncol. 2014, 120, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Miyamori, H.; Kita, D.; Takahashi, T.; Yamashita, J.; Sato, H.; Miura, R.; Yamaguchi, Y.; Okada, Y. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005, 110, 239–246. [Google Scholar] [CrossRef]
- Ohnishi, T.; Hiraga, S.; Izumoto, S.; Matsumura, H.; Kanemura, Y.; Arita, N.; Hayakawa, T. Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: Clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin. Exp. Metastasis 1998, 16, 729–741. [Google Scholar] [CrossRef]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin Promotes the Malignancy of Glioma Stem-Like Cells Via Modulation of Cell Adhesion, Differentiation, Proliferation and Chemoresistance. Front. Mol. Neurosci. 2018, 11, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, V.; Gutierrez, O.; Talamillo, A.; Velasquez, C.; Fernandez-Luna, J.L. Glioblastoma invasion factor ODZ1 is induced by microenvironmental signals through activation of a Stat3-dependent transcriptional pathway. Sci. Rep. 2021, 11, 16196. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.S.; Huang, P.H. The pathobiology of collagens in glioma. Mol. Cancer Res. 2013, 11, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pointer, K.B.; Clark, P.A.; Schroeder, A.B.; Salamat, M.S.; Eliceiri, K.W.; Kuo, J.S. Association of collagen architecture with glioblastoma patient survival. J. Neurosurg. 2017, 126, 1812–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef] [Green Version]
- Motegi, H.; Kamoshima, Y.; Terasaka, S.; Kobayashi, H.; Houkin, K. Type 1 collagen as a potential niche component for CD133-positive glioblastoma cells. Neuropathology 2014, 34, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The Tumorigenic Roles of the Cellular REDOX Regulatory Systems. Oxid. Med. Cell Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Zhong, H.; Scalzitti, J.M.; Laughner, E.; Simons, J.W.; Semenza, G.L. Expression of hypoxia-inducible factor 1alpha in brain tumors: Association with angiogenesis, invasion, and progression. Cancer 2000, 88, 2606–2618. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nakagawa, K.; Harada, H.; Nagato, S.; Furukawa, K.; Teraoka, M.; Seno, T.; Oka, K.; Iwata, S.; Ohnishi, T. Silencing hypoxia-inducible factor-1alpha inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int. J. Oncol. 2007, 30, 793–802. [Google Scholar]
- Méndez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Johansson, E.; Grassi, E.S.; Pantazopoulou, V.; Tong, B.; Lindgren, D.; Berg, T.J.; Pietras, E.J.; Axelson, H.; Pietras, A. CD44 Interacts with HIF-2α to Modulate the Hypoxic Phenotype of Perinecrotic and Perivascular Glioma Cells. Cell Rep. 2017, 20, 1641–1653. [Google Scholar] [CrossRef] [Green Version]
- Chédeville, A.L.; Lourdusamy, A.; Monteiro, A.R.; Hill, R.; Madureira, P.A. Investigating Glioblastoma Response to Hypoxia. Biomedicines 2020, 8, 310. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabilsi, N.H.; Kladde, M.P.; Suslov, O.; Brabletz, S.; Brabletz, T.; Reynolds, B.A.; Steindler, D.A. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell. Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; Den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Nordfors, K.; Haapasalo, J.; Mäkelä, K.; Granberg, K.J.; Nykter, M.; Korja, M.; Paavonen, T.; Haapasalo, H.; Soini, Y. Twist predicts poor outcome of patients with astrocytic glioma. J. Clin. Pathol. 2015, 68, 905–912. [Google Scholar] [CrossRef]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.C.; Luo, S.J.; Lin, C.L.; Chang, P.J.; Chen, M.F. Modulation of p75 neurotrophin receptor under hypoxic conditions induces migration and invasion of C6 glioma cells. Clin. Exp. Metastasis 2015, 32, 73–81. [Google Scholar] [CrossRef]
- Srivastava, C.; Irshad, K.; Dikshit, B.; Chattopadhyay, P.; Sarkar, C.; Gupta, D.K.; Sinha, S.; Chosdol, K. FAT1 modulates EMT and stemness genes expression in hypoxic glioblastoma. Int. J. Cancer 2018, 142, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, C.; Mansouri, S.; Gutiérrez, O.; Mamatjan, Y.; Mollinedo, P.; Karimi, S.; Singh, O.; Terán, N.; Martino, J.; Zadeh, G.; et al. Hypoxia Can Induce Migration of Glioblastoma Cells Through a Methylation-Dependent Control of ODZ1 Gene Expression. Front. Oncol. 2019, 10, 1036. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 683276. [Google Scholar] [CrossRef]
- Liang, Y.; Han, H.; Liu, L.; Duan, Y.; Yang, X.; Ma, C.; Zhu, Y.; Han, J.; Li, X.; Chen, Y. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis 2018, 7, 98. [Google Scholar] [CrossRef] [Green Version]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Aspects Med. 2017, 55, 140–151. [Google Scholar] [CrossRef]
- Passos, D.F.; Bernardes, V.M.; Da Silva, J.L.G.; Schetinger, M.R.C.; Leal, D.B.R. Adenosine signaling and adenosine deaminase regulation of immune responses: Impact on the immunopathogenesis of HIV infection. Purinergic Signal 2018, 14, 309–320. [Google Scholar] [CrossRef]
- Sperlágh, B.; Vizi, E.S. The role of extracellular adenosine in chemical neurotransmission in the hippocampus and Basal Ganglia: Pharmacological and clinical aspects. Curr. Top. Med. Chem. 2011, 11, 1034–1046. [Google Scholar] [CrossRef] [Green Version]
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melani, A.; De Micheli, E.; Pinna, G.; Alfieri, A.; Corte, L.D.; Pedata, F. Adenosine extracellular levels in human brain gliomas: An intraoperative microdialysis study. Neurosci. Lett. 2003, 346, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, C.; Garrido, W.; Oyarzún, C.; Fernández, K.; Segura, R.; Melo, R.; Casanello, P.; Sobrevia, L.; San Martín, R. 5’-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J. Cell Physiol. 2013, 228, 602–608. [Google Scholar] [CrossRef]
- Liu, T.Z.; Wang, X.; Bai, Y.F.; Liao, H.Z.; Qiu, S.C.; Yang, Y.Q.; Yan, X.H.; Chen, J.; Guo, H.B.; Zhang, S.Z. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int. J. Biochem. Cell Biol. 2014, 49, 8–16. [Google Scholar] [CrossRef]
- Eltzschig, H.K. Extracellular adenosine signaling in molecular medicine. J. Mol. Med. 2013, 91, 141–146. [Google Scholar] [CrossRef]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Müller, C.E. A2A and A2B adenosine receptors: The extracellular loop 2 determines high (A2A) or low affinity (A2B) for adenosine. Biochem. Pharmacol. 2020, 172, 113718. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Bozarov, A.; Wang, Y.Z.; Yu, J.G.; Wunderlich, J.; Hassanain, H.H.; Alhaj, M.; Cooke, H.J.; Grants, I.; Ren, T.; Christofi, F.L. Activation of adenosine low-affinity A3 receptors inhibits the enteric short interplexus neural circuit triggered by histamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1147–G1162. [Google Scholar] [CrossRef] [Green Version]
- Rivkees, S.A.; Thevananther, S.; Hao, H. Are A3 adenosine receptors expressed in the brain? Neuroreport 2000, 11, 1025–1230. [Google Scholar] [CrossRef]
- Cappellari, A.R.; Rockenbach, L.; Dietrich, F.; Clarimundo, V.; Glaser, T.; Braganhol, E.; Abujamra, A.L.; Roesler, R.; Ulrich, H.; Battastini, A.M. Characterization of ectonucleotidases in human medulloblastoma cell lines: Ecto-5’NT/CD73 in metastasis as potential prognostic factor. PLoS ONE 2012, 7, e47468. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [Green Version]
- Bavaresco, L.; Bernardi, A.; Braganhol, E.; Cappellari, A.R.; Rockenbach, L.; Farias, P.F.; Wink, M.R.; Delgado-Cañedo, A.; Battastini, A.M. The role of ecto-5’-nucleotidase/CD73 in glioma cell line proliferation. Mol. Cell Biochem. 2008, 319, 61–68. [Google Scholar] [CrossRef]
- Khayami, R.; Toroghian, Y.; Bahreyni, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Ebrahimi, S.; Soleimani, A.; Fiuji, H.; Avan, A.; et al. Role of adenosine signaling in the pathogenesis of head and neck cancer. J. Cell Biochem. 2018, 119, 7905–7912. [Google Scholar] [CrossRef]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef] [Green Version]
- Azambuja, J.H.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Fernandes, M.C.; Figueiró, F.; Battastini, A.M.O.; Scholl, J.N.; De Oliveira, F.H.; Spanevello, R.M.; et al. CD73 Downregulation Decreases In Vitro and In Vivo Glioblastoma Growth. Mol. Neurobiol. 2019, 56, 3260–3279. [Google Scholar] [CrossRef]
- Tsiampali, J.; Neumann, S.; Giesen, B.; Koch, K.; Maciaczyk, D.; Janiak, C.; Hänggi, D.; Maciaczyk, J. Enzymatic Activity of CD73 Modulates Invasion of Gliomas via Epithelial-Mesenchymal Transition-Like Reprogramming. Pharmaceuticals 2020, 13, 378. [Google Scholar] [CrossRef]
- Gessi, S.; Sacchetto, V.; Fogli, E.; Merighi, S.; Varani, K.; Baraldi, P.G.; Tabrizi, M.A.; Leung, E.; Maclennan, S.; Borea, P.A. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem. Pharmacol. 2010, 79, 1483–1495. [Google Scholar] [CrossRef]
- Synowitz, M.; Glass, R.; Färber, K.; Markovic, D.; Kronenberg, G.; Herrmann, K.; Schnermann, J.; Nolte, C.; Van Rooijen, N.; Kiwit, J.; et al. A1 adenosine receptors in microglia control glioblastoma-host interaction. Cancer Res. 2006, 66, 8550–8557. [Google Scholar] [CrossRef] [Green Version]
- Kitabatake, K.; Kaji, T.; Tsukimoto, M. Involvement of CD73 and A2B Receptor in Radiation-Induced DNA Damage Response and Cell Migration in Human Glioblastoma A172 Cells. Biol. Pharm. Bull. 2021, 44, 197–210. [Google Scholar] [CrossRef]
- Erices, J.I.; Niechi, I.; Uribe-Ojeda, A.; Toro, M.L.Á.; García-Romero, N.; Carrión-Navarro, J.; Monago-Sánchez, Á.; Ayuso-Sacido, Á.; San Martin, R.; Quezada-Monrás, C. The low affinity A2B adenosine receptor enhances migratory and invasive capacity in vitro and angiogenesis in vivo of glioblastoma stem-like cells. Front. Oncol. 2022, 12, 969993. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A₃ Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, Á.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D.; et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Niechi, I.; Uribe-Ojeda, A.; Erices, J.I.; Torres, Á.; Uribe, D.; Rocha, J.D.; Silva, P.; Richter, H.G.; San Martín, R.; Quezada, C. Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness. Cells 2019, 8, 1353. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem. Pharmacol. 2006, 72, 19–31. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine receptors and cancer. Handb. Exp. Pharmacol. 2009, 193, 399–441. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. S2), 1–105. [Google Scholar] [CrossRef]
- García-García, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-κB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhong, K.; Wang, Z.; Zhang, Z.; Tang, X.; Tong, A.; Zhou, L. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front. Immunol. 2022, 13, 964898. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Aasland, D.; Götzinger, L.; Hauckm, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-κB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Fletcher-Sananikone, E.; Kanji, S.; Tomimatsu, N.; Di Cristofaro, L.F.M.; Kollipara, R.K.; Saha, D.; Floyd, J.R.; Sung, P.; Hromas, R.; Burns, T.C.; et al. Elimination of Radiation-Induced Senescence in the Brain Tumor Microenvironment Attenuates Glioblastoma Recurrence. Cancer Res. 2021, 81, 5935–5947. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Symons, M. Signaling determinants of glioma cell invasion. Adv. Exp. Med. Biol. 2013, 986, 121–141. [Google Scholar] [CrossRef]
- Mentlein, R.; Hattermann, K.; Held-Feindt, J. Lost in disruption: Role of proteases in glioma invasion and progression. Biochim. Biophys. Acta 2012, 1825, 178–185. [Google Scholar] [CrossRef]
- Jo, Y.; Hwang, S.H.; Jang, J. Employing Extracellular Matrix-Based Tissue Engineering Strategies for Age-Dependent Tissue Degenerations. Int. J. Mol. Sci. 2021, 22, 9367. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [Green Version]
- Putra, V.D.L.; Kilian, K.A.; Knothe Tate, M.L. Biomechanical, biophysical, and biochemical modulators of cytoskeletal remodelling and emergent stem cell lineage commitment. Commun. Biol. 2023, 6, 75. [Google Scholar] [CrossRef]
- Ratliff, M.; Kim, H.; Qi, H.; Kim, M.; Ku, B.; Azorin, D.D.; Hausmann, D.; Khajuria, R.K.; Patel, A.; Maier, E.; et al. Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6572. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Hau, A.C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020, 10, 604121. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, L.; Luo, L.; Shu, L.; Si, X.; Chen, Z.; Xia, W.; Huang, J.; Liu, Y.; Shao, A.; et al. Opportunities and challenges of glioma organoids. Cell. Commun. Signal. 2021, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Ming, G.L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer. Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.; Knoblichm, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2013, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.J.; Shakya, S.; Barnett, A.; Wallace, L.C.; Jeon, H.; Sloan, A.; Recinos, V.; Hubert, C.G. Three-dimensional organoid culture unveils resistance to clinical therapies in adult and pediatric glioblastoma. Transl. Oncol. 2022, 15, 101251. [Google Scholar] [CrossRef]
- Darrigues, E.; Zhao, E.H.; De Loose, A.; Lee, M.P.; Borrelli, M.J.; Eoff, R.L.; Galileo, D.S.; Penthala, N.R.; Crooks, P.A.; Rodriguez, A. Biobanked Glioblastoma Patient-Derived Organoids as a Precision Medicine Model to Study Inhibition of Invasion. Int. J. Mol. Sci. 2021, 22, 10720. [Google Scholar] [CrossRef]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of pa-tient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvi-ronment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell. Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell. Rep. 2018, 26, 3203–3211. [Google Scholar]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020, 31, 107738. [Google Scholar] [CrossRef]



| Source | Molecule | Mechanism | Reference |
|---|---|---|---|
| Extracellular matrix | HA |
| [80,81] |
| OPN |
| [82] | |
| TNC |
| [84,85,86,87,88,89,90] | |
| Laminin-8 |
| [91,92] | |
| Laminin-2 and -5 |
| [93,94,95] | |
| BCAN |
| [96,97] | |
| FN |
| [98,99,100,101,102] | |
| Collagen |
| [103,104,105] |
| Source | Molecule | Mechanism | Reference |
|---|---|---|---|
| Hypoxia | HIF-1α |
| [115,116,117] |
| HIF-2α |
| [118,119] | |
| PAI-1, S100A10, uPAR |
| [120] | |
| EMT markers |
| [121,122] | |
| ZEB1 |
| [123,124,125] | |
| Twist1 |
| [126,127,128] | |
| FAT1 |
| [129] | |
| ODZ1 |
| [131] |
| Source | Protein | Mechanisms | Reference |
|---|---|---|---|
| Adenosine | CD73 |
| [155,156,157] |
| A3AR |
| [158,162,163] | |
| A1AR |
| [159] | |
| A2BAR |
| [160,161] | |
| PAP |
| [145,163] | |
| ADA |
| [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erices, J.I.; Bizama, C.; Niechi, I.; Uribe, D.; Rosales, A.; Fabres, K.; Navarro-Martínez, G.; Torres, Á.; San Martín, R.; Roa, J.C.; et al. Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 7047. https://doi.org/10.3390/ijms24087047
Erices JI, Bizama C, Niechi I, Uribe D, Rosales A, Fabres K, Navarro-Martínez G, Torres Á, San Martín R, Roa JC, et al. Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets. International Journal of Molecular Sciences. 2023; 24(8):7047. https://doi.org/10.3390/ijms24087047
Chicago/Turabian StyleErices, José Ignacio, Carolina Bizama, Ignacio Niechi, Daniel Uribe, Arnaldo Rosales, Karen Fabres, Giovanna Navarro-Martínez, Ángelo Torres, Rody San Martín, Juan Carlos Roa, and et al. 2023. "Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets" International Journal of Molecular Sciences 24, no. 8: 7047. https://doi.org/10.3390/ijms24087047