
Evolution-Informed Strategies for Combating Drug Resistance in Cancer

{kind=link}
{kind=link}
{kind=link}
Abstract
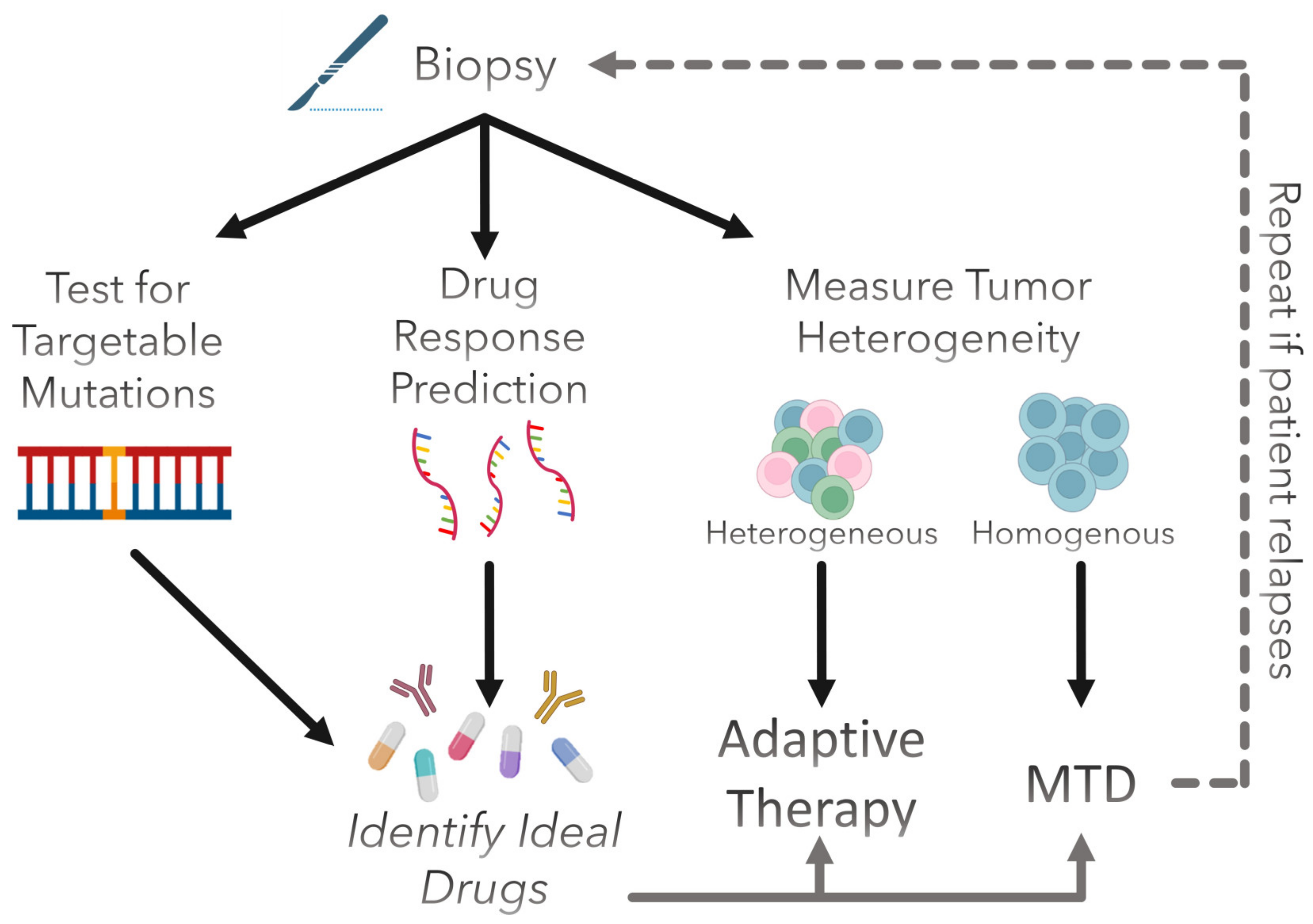
:1. Introduction
2. Predicting Drug Sensitivity to Improve Treatment Planning
2.1. Gene Signatures
2.2. Computational Models
2.3. Experimental Evolution and Collateral Sensitivity
3. Controlling Evolution by Exploiting Ecological Dynamics
Adaptive Therapy
4. Clinical Prospect of Evolution-Informed Treatment Strategies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Cole, S.; Bhardwaj, G.; Gerlach, J.H.; Mackie, E.J.; Grant, E.C.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Shen, D.-W.; Goldenberg, S.; Pastan, I.; Gottesman, M.M. Decreased accumulation of [14c]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J. Cell. Physiol. 2000, 183, 108–116. [Google Scholar] [CrossRef]
- Altan, N.; Chen, Y.; Schindler, M.; Simon, S.M. Defective Acidification in Human Breast Tumor Cells and Implications for Chemotherapy. J. Exp. Med. 1998, 187, 1583–1598. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009, 10, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.L.; Kümler, I.; Palshof, J.A.; Andersson, M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast 2013, 22, 1–12. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [Green Version]
- Mounier, N.; Briere, J.; Gisselbrecht, C.; Emile, J.-F.; Lederlin, P.; Sebban, C.; Berger, F.; Bosly, A.; Morel, P.; Tilly, H.; et al. Rituximab plus CHOP (R-CHOP) overcomes bcl-2–associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood 2003, 101, 4279–4284. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.; Malempati, S. Ewing sarcoma in adolescents and young adults: Diagnosis and treatment. Clin. Oncol. Adolesc. Young Adults 2014, 2014, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [Green Version]
- Farrokhian, N.; Maltas, J.; Dinh, M.; Durmaz, A.; Ellsworth, P.; Hitomi, M.; McClure, E.; Marusyk, A.; Kaznatcheev, A.; Scott, J.G. Measuring competitive exclusion in non–small cell lung cancer. Sci. Adv. 2022, 8, eabm7212. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Dinh, M.N.; Brown, J.S.; Zhang, J.; Anderson, A.R.; Gatenby, R.A. Multidrug Cancer Therapy in Metastatic Castrate-Resistant Prostate Cancer: An Evolution-Based Strategy. Clin. Cancer Res. 2019, 25, 4413–4421. [Google Scholar] [CrossRef] [Green Version]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Pomeroy, A.E.; Schmidt, E.V.; Sorger, P.K.; Palmer, A.C. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer 2022, 8, 915–929. [Google Scholar] [CrossRef]
- Roell, K.R.; Reif, D.M.; Motsinger-Reif, A.A. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from Across Disciplines. Front. Pharmacol. 2017, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Zöllner, S.; Amatruda, J.; Bauer, S.; Collaud, S.; De Álava, E.; DuBois, S.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S. Hospital antibiogram: A necessity. Indian J. Med. Microbiol. 2010, 28, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Gopal, P.; Kuzmishin, G.B.; DeBernardo, R.; Koyfman, S.A.; Jha, B.K.; Mian, O.Y.; Scott, J.; Adams, D.J.; Peacock, C.D.; et al. Case study: Patient-derived clear cell adenocarcinoma xenograft model longitudinally predicts treatment response. NPJ Precis. Oncol. 2018, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.; Tang, G.; Shak, S.; Kim, C.; Baker, J.; Kim, W.; Cronin, M.; Baehner, F.L.; Watson, D.; Bryant, J.; et al. Gene Expression and Benefit of Chemotherapy in Women with Node-Negative, Estrogen Receptor–Positive Breast Cancer. J. Clin. Oncol. 2006, 24, 3726–3734. [Google Scholar] [CrossRef]
- Soliman, H.; Shah, V.; Srkalovic, G.; Mahtani, R.; Levine, E.; Mavromatis, B.; Srinivasiah, J.; Kassar, M.; Gabordi, R.; Qamar, R.; et al. MammaPrint guides treatment decisions in breast Cancer: Results of the IMPACt trial. BMC Cancer 2020, 20, 81. [Google Scholar] [CrossRef] [Green Version]
- Jairath, N.K.; Dal Pra, A.; Vince, R., Jr.; Dess, R.T.; Jackson, W.C.; Tosoian, J.J.; McBride, S.M.; Zhao, S.G.; Berlin, A.; Mahal, B.A.; et al. A systematic review of the evidence for the decipher genomic classifier in prostate cancer. Eur. Urol. 2021, 79, 374–383. [Google Scholar] [CrossRef]
- Eschrich, S.A.; Pramana, J.; Zhang, H.; Zhao, H.; Boulware, D.; Lee, J.-H.; Bloom, G.; Rocha-Lima, C.; Kelley, S.; Calvin, D.P.; et al. A Gene Expression Model of Intrinsic Tumor Radiosensitivity: Prediction of Response and Prognosis After Chemoradiation. Int. J. Radiat. Oncol. 2009, 75, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Eschrich, S.; Zhang, H.; Zhao, H.; Boulware, D.; Lee, J.-H.; Bloom, G.; Torres-Roca, J.F. Systems Biology Modeling of the Radiation Sensitivity Network: A Biomarker Discovery Platform. Int. J. Radiat. Oncol. 2009, 75, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Eschrich, S.A.; Fulp, W.J.; Pawitan, Y.; Foekens, J.A.; Smid, M.; Martens, J.W.; Echevarria, M.; Kamath, V.; Lee, J.-H.; Harris, E.E.; et al. Validation of a Radiosensitivity Molecular Signature in Breast Cancer. Clin. Cancer Res. 2012, 18, 5134–5143. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.A.; Chinnaiyan, P.; Fulp, W.J.; Eschrich, S.; Torres-Roca, J.F.; Caudell, J.J. The radiosensitivity index predicts for overall survival in glioblastoma. Oncotarget 2015, 6, 34414–34422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.A.; Scott, J.G.; Arrington, J.A.; Naghavi, A.O.; Grass, G.D.; Perez, B.A.; Caudell, J.J.; Berglund, A.E.; Welsh, E.A.; Eschrich, S.A.; et al. Radiosensitivity of Lung Metastases by Primary Histology and Implications for Stereotactic Body Radiation Therapy Using the Genomically Adjusted Radiation Dose. J. Thorac. Oncol. 2018, 13, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Roca, J.F.; Fulp, W.J.; Caudell, J.J.; Servant, N.; Bollet, M.A.; van de Vijver, M.; Naghavi, A.O.; Harris, E.E.; Eschrich, S.A. Integration of a Radiosensitivity Molecular Signature Into the Assessment of Local Recurrence Risk in Breast Cancer. Int. J. Radiat. Oncol. 2015, 93, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, T.; Hoffe, S.E.; Fulp, W.; Frakes, J.; Coppola, D.; Springett, G.M.; Malafa, M.P.; Harris, C.L.; Eschrich, S.A.; Torres-Roca, J.F.; et al. Radiosensitivity index predicts for survival with adjuvant radiation in resectable pancreatic cancer. Radiother. Oncol. 2015, 117, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.G.; Berglund, A.; Schell, M.J.; Mihaylov, I.; Fulp, W.J.; Yue, B.; Welsh, E.; Caudell, J.J.; Ahmed, K.; Strom, T.S.; et al. A genome-based model for adjusting radiotherapy dose (GARD): A retrospective, cohort-based study. Lancet Oncol. 2016, 18, 202–211. [Google Scholar] [CrossRef]
- Strom, T.; Torres-Roca, J.F.; Parekh, A.; Naghavi, A.O.; Caudell, J.J.; Oliver, D.E.; Messina, J.L.; Khushalani, N.I.; Zager, J.S.; Sarnaik, A.; et al. Regional Radiation Therapy Impacts Outcome for Node-Positive Cutaneous Melanoma. J. Natl. Compr. Cancer Netw. 2017, 15, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, M.; Staaf, J.; Edén, P.; Wärnberg, F.; Bergh, J.; Malmström, P.; Fernö, M.; Niméus, E.; Fredriksson, I. Identification and validation of single-sample breast cancer radiosensitivity gene expression predictors. Breast Cancer Res. 2018, 20, 64. [Google Scholar] [CrossRef]
- Locati, L.D.; Serafini, M.S.; Iannò, M.F.; Carenzo, A.; Orlandi, E.; Resteghini, C.; Cavalieri, S.; Bossi, P.; Canevari, S.; Licitra, L.; et al. Mining of Self-Organizing Map Gene-Expression Portraits Reveals Prognostic Stratification of HPV-Positive Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, H.; Prince, A.; Figura, N.B.; Peacock, J.S.; Fernandez, D.C.; Montejo, M.E.; Chon, H.S.; Wenham, R.M.; Eschrich, S.A.; Torres-Roca, J.F.; et al. Using the Radiosensitivity Index (RSI) to Predict Pelvic Failure in Endometrial Cancer Treated with Adjuvant Radiation Therapy. Int. J. Radiat. Oncol. 2019, 106, 496–502. [Google Scholar] [CrossRef]
- Thiruthaneeswaran, N.; Bibby, B.; Pereira, R.; More, E.; Denley, H.; Henry, A.; Wylie, J.; Hoskin, P.; Bristow, R.; Choudhury, A.; et al. OC-1031: The radiosensitivity index predicts benefit from HDR brachytherapy in high-risk prostate cancer. Radiother. Oncol. 2020, 152, S1086–S1087. [Google Scholar] [CrossRef]
- Scott, J.G.; Sedor, G.; Scarborough, J.; Kattan, M.W.; Peacock, J.; Grass, G.D.; Mellon, E.A.; Thapa, R.; Schell, M.; Waller, A.; et al. Personalizing Radiotherapy Prescription Dose Using Genomic Markers of Radiosensitivity and Normal Tissue Toxicity in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2020, 16, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G.; Sedor, G.; Ellsworth, P.; Scarborough, A.J.; Ahmed, A.K.; Oliver, E.D.; Eschrich, A.S.; Kattan, M.W.; Torres-Roca, J.F. Pan-cancer prediction of radiotherapy benefit using genomic-adjusted radiation dose (GARD): A cohort-based pooled analysis. Lancet Oncol. 2021, 22, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, J.A.; Eschrich, S.A.; Torres-Roca, J.; Dhawan, A.; Scott, J.G. Exploiting convergent evolution to derive a pan-cancer cisplatin sensitivity gene expression signature. medRxiv 2021. [Google Scholar] [CrossRef]
- Scarborough, J.A.; McClure, E.; Anderson, P.; Dhawan, A.; Durmaz, A.; Lessnick, S.L.; Hitomi, M.; Scott, J.G. Identifying States of Collateral Sensitivity during the Evolution of Therapeutic Resistance in Ewing’s Sarcoma. iScience 2020, 23, 101293. [Google Scholar] [CrossRef]
- Meza-Junco, J.; Sawyer, M.B. Drug exposure: Still an excellent biomarker. Biomarkers Med. 2009, 3, 723–731. [Google Scholar] [CrossRef]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.M.; Rizzo, S.; van der Zee, A.; Plumb, A.J.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef] [Green Version]
- Sakellaropoulos, T.; Vougas, K.; Narang, S.; Koinis, F.; Kotsinas, A.; Polyzos, A.; Moss, T.J.; Piha-Paul, S.; Zhou, H.; Kardala, E.; et al. A Deep Learning Framework for Predicting Response to Therapy in Cancer. Cell Rep. 2019, 29, 3367–3373.e4. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.; Rockstroh, A.; Lehman, M.; Ratther, E.; Jain, A.; Anand, A.; Gupta, A.; Bhattacharya, N.; Poonia, S.; Rai, P.; et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022, 13, 5680. [Google Scholar] [CrossRef]
- Suphavilai, C.; Chia, S.; Sharma, A.; Tu, L.; Da Silva, R.P.; Mongia, A.; DasGupta, R.; Nagarajan, N. Predicting heterogeneity in clone-specific therapeutic vulnerabilities using single-cell transcriptomic signatures. Genome Med. 2021, 13, 189. [Google Scholar] [CrossRef]
- Elemento, O.; Leslie, C.; Lundin, J.; Tourassi, G. Artificial intelligence in cancer research, diagnosis and therapy. Nat. Rev. Cancer 2021, 21, 747–752. [Google Scholar] [CrossRef]
- Rees, M.G.; Brenan, L.; Carmo, M.D.; Duggan, P.; Bajrami, B.; Arciprete, M.; Boghossian, A.; Vaimberg, E.; Ferrara, S.J.; Lewis, T.A.; et al. Systematic identification of biomarker-driven drug combinations to overcome resistance. Nat. Chem. Biol. 2022, 18, 615–624. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Courtiol, P.; Maussion, C.; Moarii, M.; Pronier, E.; Pilcer, S.; Sefta, M.; Manceron, P.; Toldo, S.; Zaslavskiy, M.; Le Stang, N.; et al. Deep learning-based classification of mesothelioma improves prediction of patient outcome. Nat. Med. 2019, 25, 1519–1525. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Park, J.; Fong, S.H.; Sanchez, K.S.; Lee, J.; Kreisberg, J.F.; Ma, J.; Ideker, T. Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells. Cancer Cell 2020, 38, 672–684.e6. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jensen, M.A.; Zenklusen, J.C. A Practical Guide to The Cancer Genome Atlas (TCGA). In Statistical Genomics; Humana Press: New York, NY, USA, 2016; Volume 1418, pp. 111–141. [Google Scholar] [CrossRef]
- Lenski, E.R. Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J. 2017, 11, 2181–2194. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, K.; Przybilla, M.; Xu, H.; Kotler, E.; Karagyozova, K.; Sockell, A.; Liu, K.; Mah, A.; Lo, Y.H.; Lu, B.; et al. Experimental evolution in TP53 deficient human gastric organoids recapitulates tumorigenesis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Dhawan, A.; Nichol, D.; Kinose, F.; Abazeed, M.E.; Marusyk, A.; Haura, E.B.; Scott, J.G. Collateral sensitivity networks revealevolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep. 2017, 7, 1232. [Google Scholar] [CrossRef] [Green Version]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Vaghari-Tabari, M.; Hassanpour, P.; Sadeghsoltani, F.; Malakoti, F.; Alemi, F.; Qujeq, D.; Asemi, Z.; Yousefi, B. CRISPR/Cas9 gene editing: A new approach for overcoming drug resistance in cancer. Cell. Mol. Biol. Lett. 2022, 27, 49. [Google Scholar] [CrossRef]
- Morgan, D.; Jost, T.A.; De Santiago, C.; Brock, A. Applications of high-resolution clone tracking technologies in cancer. Curr. Opin. Biomed. Eng. 2021, 19, 100317. [Google Scholar] [CrossRef]
- Velde, R.V.; Yoon, N.; Marusyk, V.; Durmaz, A.; Dhawan, A.; Miroshnychenko, D.; Lozano-Peral, D.; Desai, B.; Balynska, O.; Poleszhuk, J.; et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat. Commun. 2020, 11, 2393. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Chauvistré, H.; Shannan, B.; Daignault-Mill, S.M.; Ju, R.J.; Picard, D.; Egetemaier, S.; Váraljai, R.; Gibhardt, C.S.; Sechi, A.; Kaschani, F.; et al. Persister state-directed transitioning and vulnerability in melanoma. Nat. Commun. 2022, 13, 3055. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Brown, J.; Vincent, T. Lessons from Applied Ecology: Cancer Control Using an Evolutionary Double Bind. Cancer Res. 2009, 69, 7499–7502. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.; Brown, J. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 8, a033415. [Google Scholar] [CrossRef] [Green Version]
- Kotler, B.P.; Brown, J.S. Cancer Community Ecology. Cancer Control. 2020, 27, 1073274820951776. [Google Scholar] [CrossRef]
- Miller, A.K.; Brown, J.S.; Enderling, H.; Basanta, D.; Whelan, C.J. The Evolutionary Ecology of Dormancy in Nature and in Cancer. Front. Ecol. Evol. 2021, 9, 676802. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518. [Google Scholar] [CrossRef]
- Dhawan, A.; Graham, T.A.; Fletcher, A.G. A Computational Modeling Approach for Deriving Biomarkers to Predict Cancer Risk in Premalignant DiseaseBiomarker Evaluation for Premalignant Disease In Silico. Cancer Prev. Res. 2016, 9, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Archetti, M.; Pienta, K.J. Cooperation among cancer cells: Applying game theory to cancer. Nat. Rev. Cancer 2018, 19, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Stanková, K.; Brown, J.S.; Dalton, W.S.; Gatenby, R.A. Optimizing cancer treatment using game theory: A review. JAMA Oncol. 2019, 5, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [CrossRef]
- Cunningham, J.; Thuijsman, F.; Peeters, R.; Viossat, Y.; Brown, J.; Gatenby, R.; Staňková, K. Optimal control to reach eco-evolutionary stability in metastatic castrate-resistant prostate cancer. PLoS ONE 2020, 15, e0243386. [Google Scholar] [CrossRef]
- Szakács, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef]
- Hansen, E.; Karslake, J.; Woods, R.J.; Read, A.F.; Wood, K.B. Antibiotics can be used to contain drug-resistant bacteria by maintaining sufficiently large sensitive populations. PLoS Biol. 2020, 18, e3000713. [Google Scholar] [CrossRef] [PubMed]
- Wargo, A.R.; Huijben, S.; De Roode, J.C.; Shepherd, J.; Read, A.F. Competitive release and facilitation of drug-resistant parasites after therapeutic chemotherapy in a rodent malaria model. Proc. Natl. Acad. Sci. USA 2007, 104, 19914–19919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijben, S.; Bell, A.S.; Sim, D.G.; Tomasello, D.; Mideo, N.; Day, T.; Read, A.F. Aggressive Chemotherapy and the Selection of Drug Resistant Pathogens. PLoS Pathog. 2013, 9, e1003578. [Google Scholar] [CrossRef] [Green Version]
- Pollitt, L.C.; Huijben, S.; Sim, D.G.; Salathé, R.M.; Jones, M.J.; Read, A.F. Rapid Response to Selection, Competitive Release and Increased Transmission Potential of Artesunate-Selected Plasmodium chabaudi Malaria Parasites. PLoS Pathog. 2014, 10, e1004019. [Google Scholar] [CrossRef]
- Kaznatcheev, A.; Peacock, J.; Basanta, D.; Marusyk, A.; Scott, J.G. Fibroblasts and alectinib switch the evolutionary games played by non-small cell lung cancer. Nat. Ecol. Evol. 2019, 3, 450–456. [Google Scholar] [CrossRef]
- Stanková, K. Resistance games. Nat. Ecol. Evol. 2019, 3, 336–337. [Google Scholar] [CrossRef] [Green Version]
- Wölfl, B.; Rietmole, H.T.; Salvioli, M.; Kaznatcheev, A.; Thuijsman, F.; Brown, J.S.; Burgering, B.; Staňková, K. The Contribution of Evolutionary Game Theory to Understanding and Treating Cancer. Dyn. Games Appl. 2021, 12, 313–342. [Google Scholar] [CrossRef]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef] [Green Version]
- Smalley, I.; Kim, E.; Li, J.; Spence, P.; Wyatt, C.J.; Eroglu, Z.; Sondak, V.K.; Messina, J.L.; Babacan, N.A.; Maria-Engler, S.; et al. Leveraging transcriptional dynamics to improve BRAF inhibitor responses in melanoma. eBioMedicine 2019, 48, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [Green Version]
- West, J.; You, L.; Zhang, J.; Gatenby, R.A.; Brown, J.S.; Newton, P.K.; Anderson, A.R. Towards Multidrug Adaptive Therapy. Cancer Res. 2020, 80, 1578–1589. [Google Scholar] [CrossRef]
- Strobl, M.A.R.; Gallaher, J.; West, J.; Robertson-Tessi, M.; Maini, P.K.; Anderson, A.R.A. Spatial structure impacts adaptive therapy by shaping intra-tumoral competition. Commun. Med. 2022, 2, 46. [Google Scholar] [CrossRef]
- Strobl, M.A.; West, J.; Viossat, Y.; Damaghi, M.; Robertson-Tessi, M.; Brown, J.S.; Gatenby, R.A.; Maini, P.K.; Anderson, A.R. Turnover Modulates the Need for a Cost of Resistance in Adaptive Therapy. Cancer Res. 2021, 81, 1135–1147. [Google Scholar] [CrossRef]
- Yoon, N.; Velde, R.V.; Marusyk, A.; Scott, J.G. Optimal Therapy Scheduling Based on a Pair of Collaterally Sensitive Drugs. Bull. Math. Biol. 2018, 80, 1776–1809. [Google Scholar] [CrossRef]
- Yoon, N.; Krishnan, N.; Scott, J. Theoretical modeling of collaterally sensitive drug cycles: Shaping heterogeneity to allow adaptive therapy. J. Math. Biol. 2021, 83, 47. [Google Scholar] [CrossRef]
- Maltas, J.; Wood, K.B. Pervasive and diverse collateral sensitivity profiles inform optimal strategies to limit antibiotic resistance. PLoS Biol. 2019, 17, e3000515. [Google Scholar] [CrossRef] [Green Version]
- Weaver, D.T.; Maltas, J.; Scott, J.G. Reinforcement Learning informs optimal treatment strategies to limit antibiotic resistance. bioRxiv 2023. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin-Rahardja, K.; Weaver, D.T.; Scarborough, J.A.; Scott, J.G. Evolution-Informed Strategies for Combating Drug Resistance in Cancer. Int. J. Mol. Sci. 2023, 24, 6738. https://doi.org/10.3390/ijms24076738
Lin-Rahardja K, Weaver DT, Scarborough JA, Scott JG. Evolution-Informed Strategies for Combating Drug Resistance in Cancer. International Journal of Molecular Sciences. 2023; 24(7):6738. https://doi.org/10.3390/ijms24076738
Chicago/Turabian StyleLin-Rahardja, Kristi, Davis T. Weaver, Jessica A. Scarborough, and Jacob G. Scott. 2023. "Evolution-Informed Strategies for Combating Drug Resistance in Cancer" International Journal of Molecular Sciences 24, no. 7: 6738. https://doi.org/10.3390/ijms24076738