
Comparative Transcriptome Analysis Reveals a Potential Regulatory Network for Ogura Cytoplasmic Male Sterility in Cabbage (Brassica oleracea L.)
 , , and
, , and
Abstract
:1. Introduction
2. Results
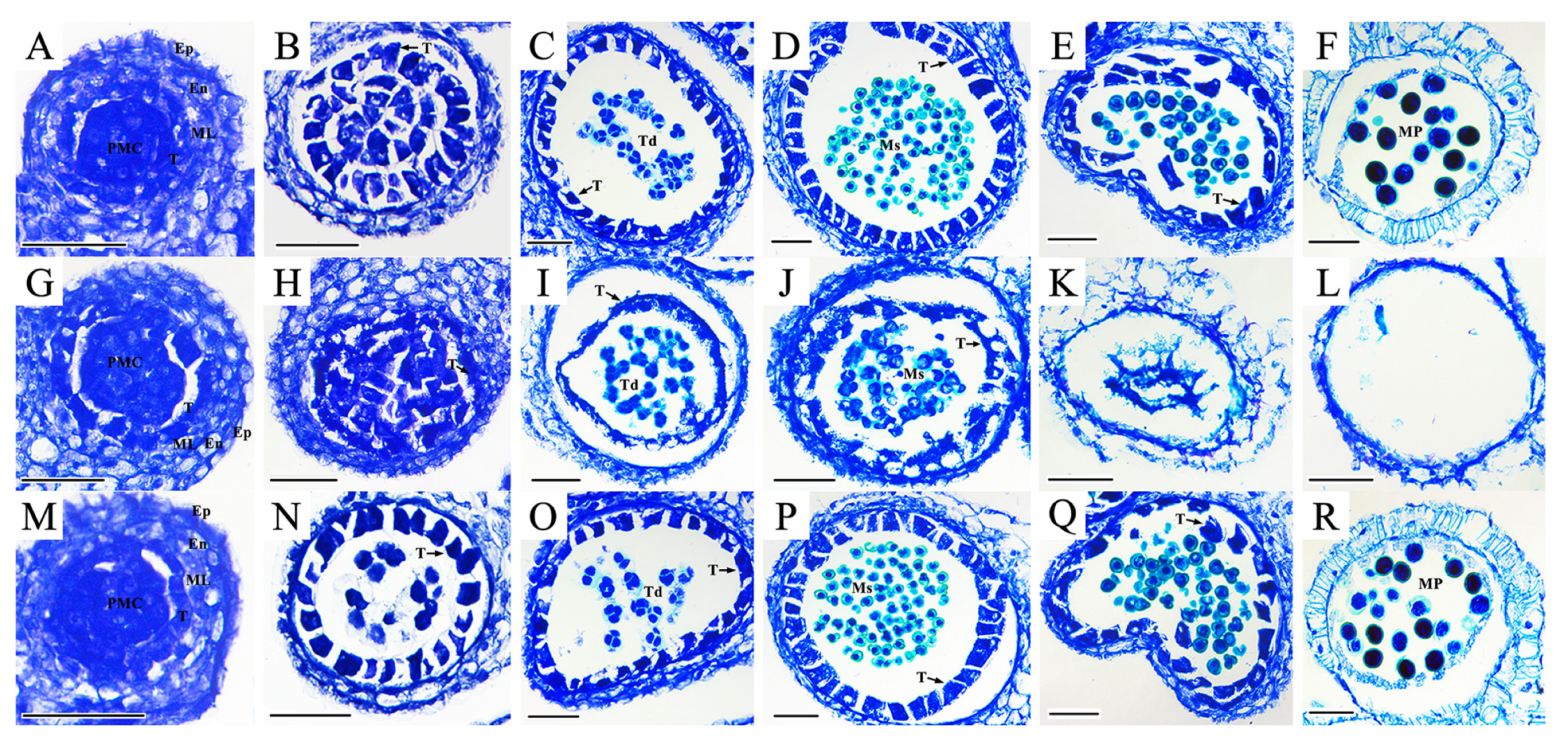
2.1. Morphological and Cytological Comparison between the Maintainer Line, Ogura CMS Line, and Restorer Line
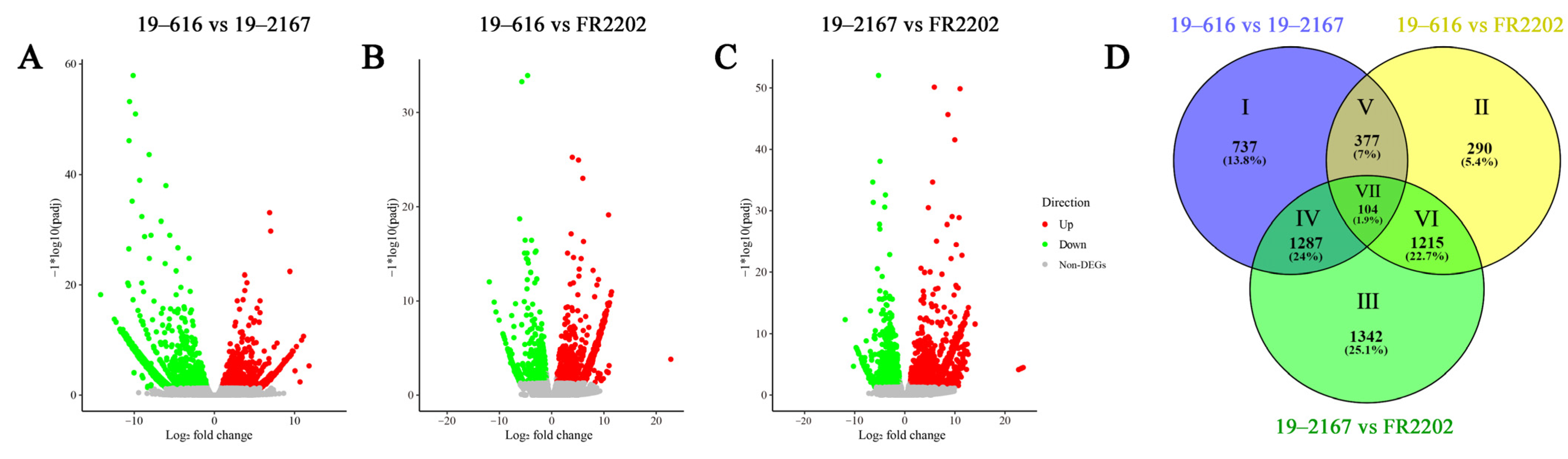
2.2. Transcriptome Sequencing and Assembly
2.3. Differential Expression Analysis of Nuclear Genes
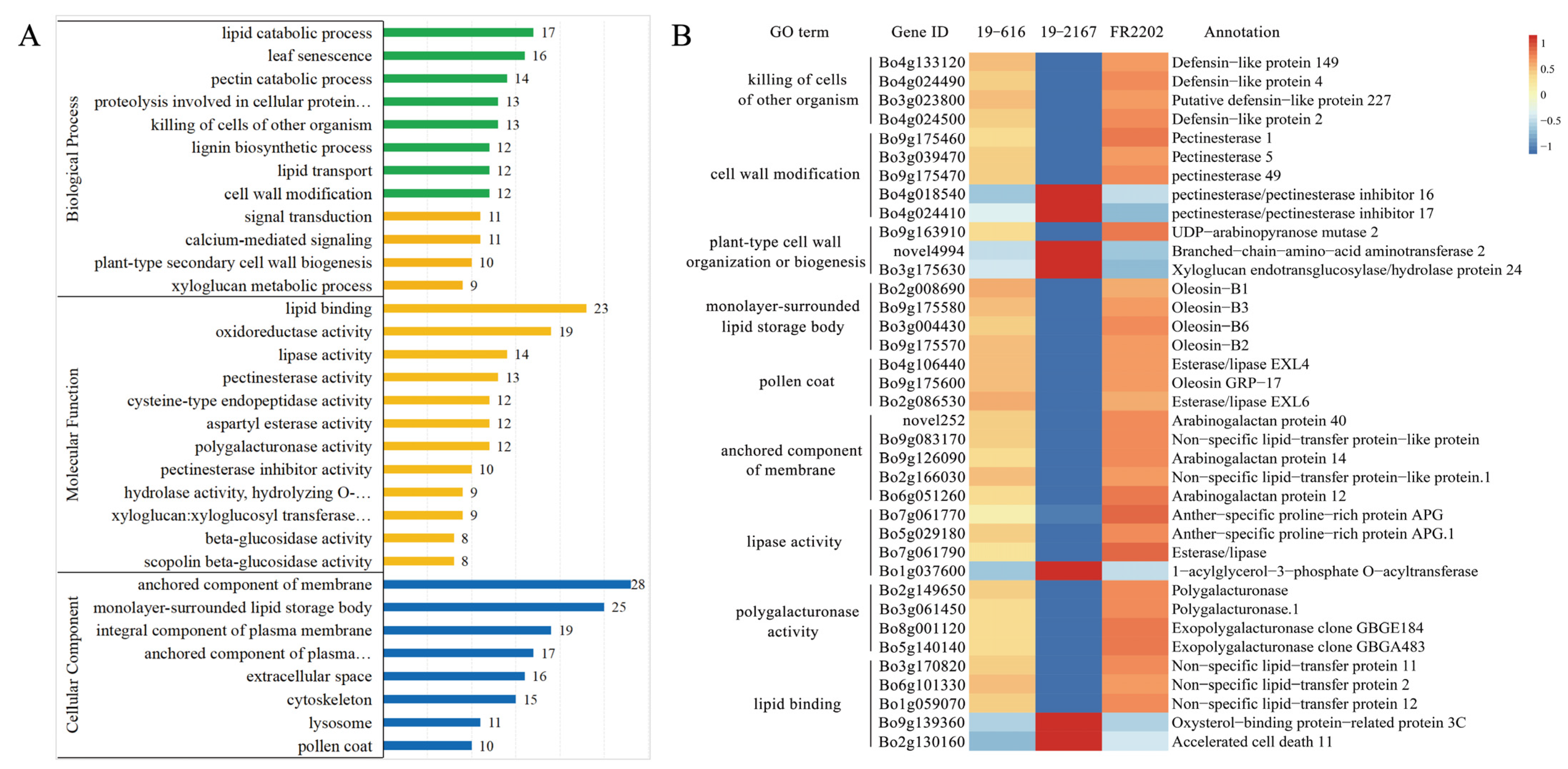
2.4. Functional Annotation and Enrichment Analysis of DEGs
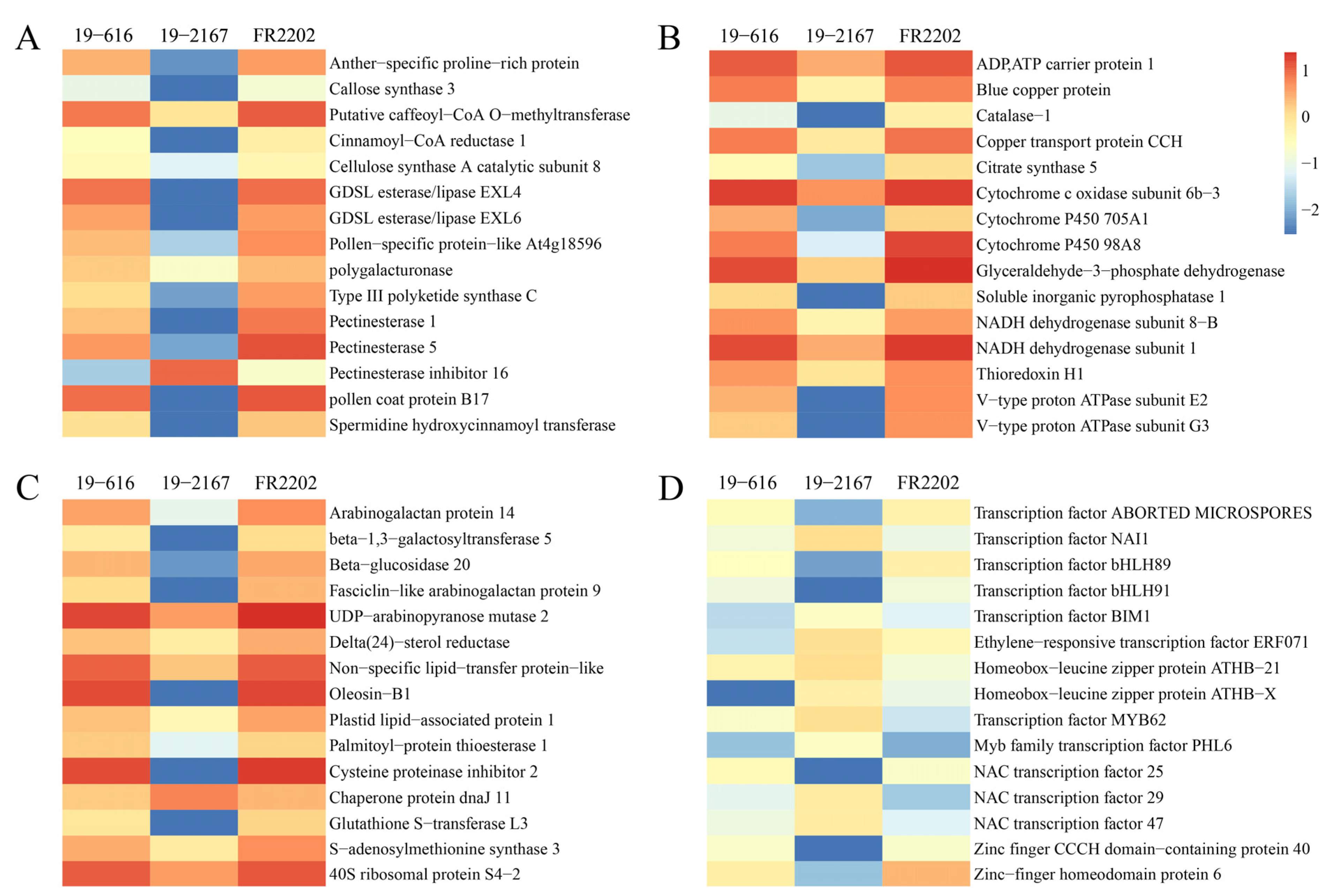
2.5. DEGs Involved in Pollen Development
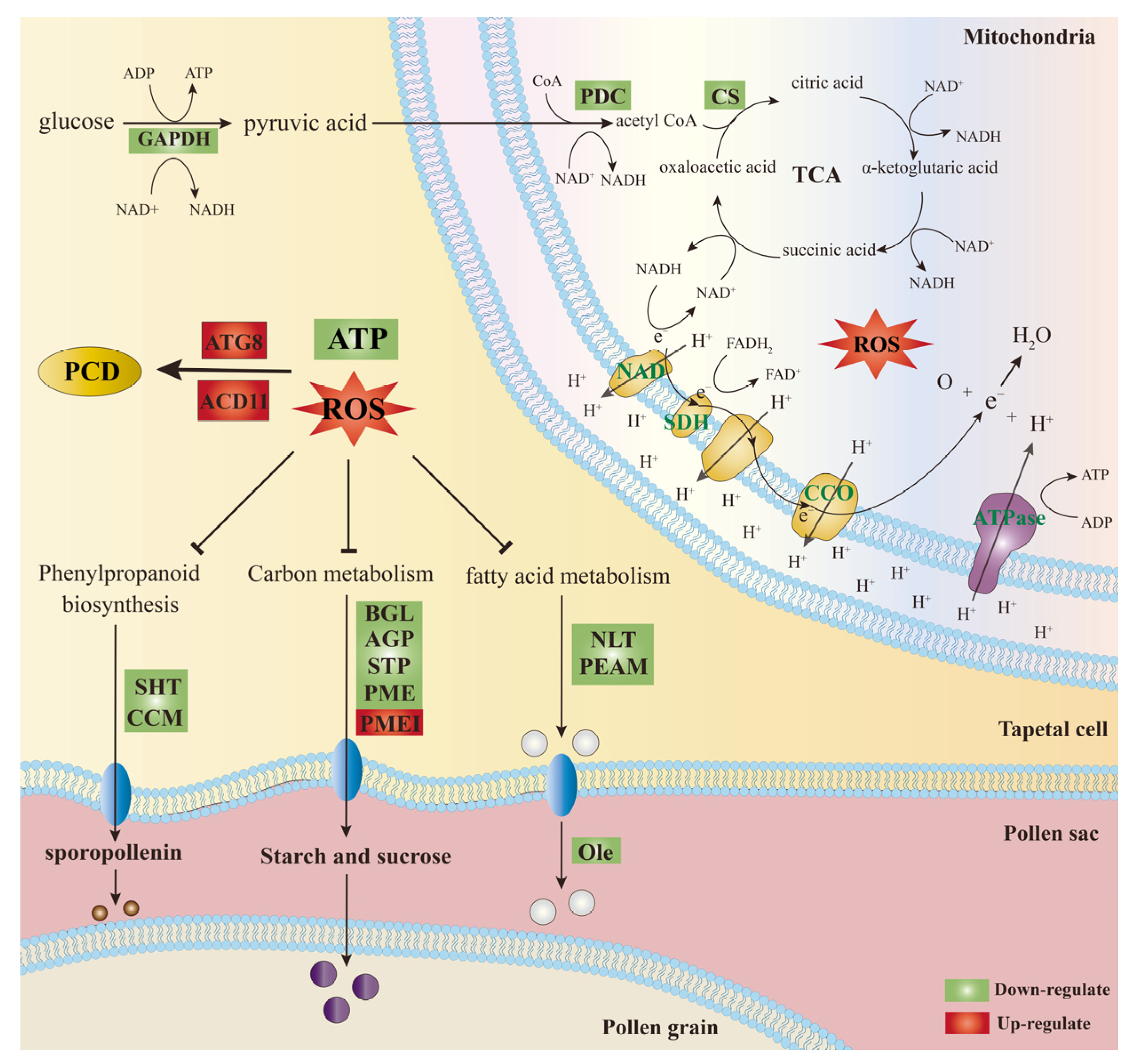
2.6. DEGs Involved in Energy Metabolism
2.7. DEGs Involved in Carbohydrate, Lipid, and Protein Metabolism
2.8. Transcription Factors among the Core DEGs
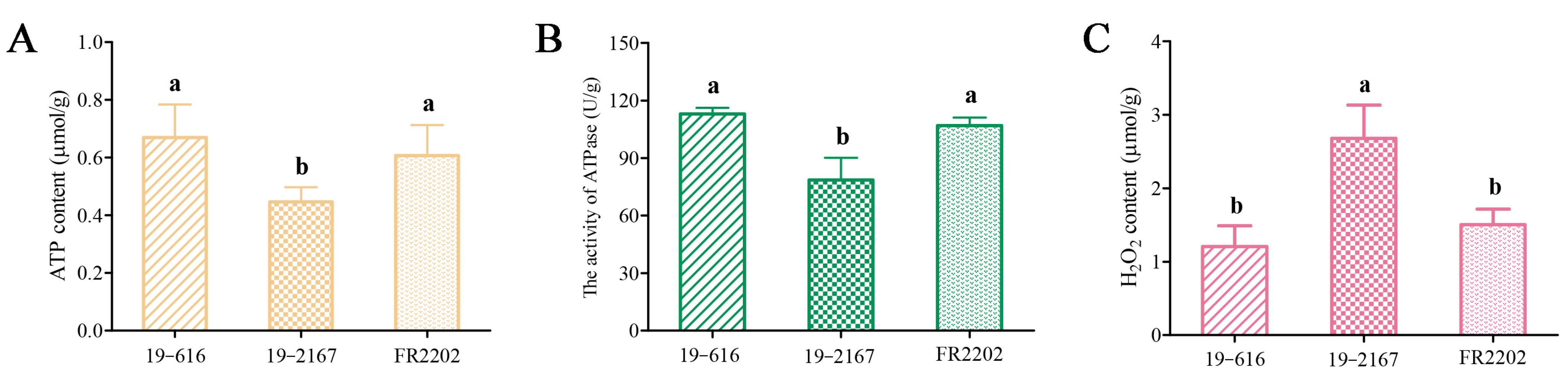
2.9. Quantification of Biochemical Indices Involved in Energy Metabolism
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Observation of Pollen Viability
4.3. Library Construction and ONT Sequencing
4.4. Sequencing Data Processing and Full-Length Transcript Identification
4.5. Quantitative Analysis and Screening of DEGs
4.6. Annotation and Enrichment Analysis of DEGs
4.7. Real-Time Quantitative PCR Analysis
4.8. Detection of Biochemical Indices Related to Energy Metabolism
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic Male Sterility |
| DEGs | Differentially expressed genes |
| PCD | Programmed cell death |
| ROS | Reactive oxygen species |
| TFs | Transcription factors |
References
- FAO. Food and Agriculture Organization of the United Nations, FAOSTAT. 2021. Available online: https://www.fao.org/faostat/zh/#data/QCL (accessed on 25 December 2022).
- Kuera, V.; Věra, C.; Miroslava, V.; Miroslav, K. Hybrid breeding of cauliflower using self-incompatibility and cytoplasmic male sterility. Hortic. Sci. (HORTSCI) 2006, 33, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.S.; Singh, N.; Bhatia, R.; Parkash, C.; Chandel, C. Genetic combining ability and heterosis for important vitamins and antioxidant pigments in cauliflower (Brassica oleracea var. botrytis L.). Euphytica 2014, 195, 169–181. [Google Scholar] [CrossRef]
- Singh, S.; Dey, S.S.; Bhatia, R.; Kumar, R.; Sharma, K.; Behera, T.K. Heterosis and combining ability in cytoplasmic male sterile and doubled haploid based Brassica oleracea progenies and prediction of heterosis using microsatellites. PLoS ONE 2019, 14, e0210772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Li, Z.; Han, F.; Zhang, B.; Li, X.; Fang, Z.; Yang, L.; Zhuang, M.; Lv, H.; Liu, Y.; et al. Utilization of Ogura CMS germplasm with the clubroot resistance gene by fertility restoration and cytoplasm replacement in Brassica oleracea L. Hortic. Res. 2020, 7, 61. [Google Scholar] [CrossRef]
- Heng, S.; Liu, S.; Xia, C.; Tang, H.; Xie, F.; Fu, T.; Wan, Z. Morphological and genetic characterization of a new cytoplasmic male sterility system (oxa CMS) in stem mustard (Brassica juncea). Theor. Appl. Genet. 2018, 131, 59–66. [Google Scholar] [CrossRef]
- Yamagishi, H.; Bhat, S.R. Cytoplasmic male sterility in Brassicaceae crops. Breed. Sci. 2014, 64, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Shinada, T.; Kikuchi, Y.; Fujimoto, R.; Kishitani, S. An alloplasmic male-sterile line of Brassica oleracea harboring the mitochondria from Diplotaxis muralis expresses a novel chimeric open reading frame, orf72. Plant Cell Physiol. 2006, 47, 549–553. [Google Scholar] [CrossRef]
- Fujii, S.; Toriyama, K. Genome barriers between nuclei and mitochondria exemplified by cytoplasmic male sterility. Plant Cell Physiol. 2008, 49, 1484–1494. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lezhneva, L.; Arnal, N.; Quadrado, M.; Mireau, H. The radish Ogura fertility restorer impedes translation elongation along its cognate CMS-causing mRNA. Proc. Natl. Acad. Sci. USA 2021, 118, e2105274118. [Google Scholar] [CrossRef]
- Ren, W.; Si, J.; Chen, L.; Fang, Z.; Zhuang, M.; Lv, H.; Wang, Y.; Ji, J.; Yu, H.; Zhang, Y. Mechanism and Utilization of Ogura Cytoplasmic Male Sterility in Cruciferae Crops. Int. J. Mol. Sci. 2022, 23, 9099. [Google Scholar] [CrossRef]
- Ogura, H. Studies of a new male-sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ. 1968, 6, 39–78. [Google Scholar]
- Bannerot, H.; Boulidard, L.; Canderon, Y.; Tempe, J. Transfer of cytoplasmic male sterility from Raphanus sativus to Brassica oleracea. Eucarpia Crucif. Newsl. 1974, 1, 52–54. [Google Scholar]
- Walters, T.W.; Mutschler, M.A.; Earle, E.D. Protoplast fusion-derived Ogura male sterile cauliflower with cold tolerance. Plant Cell Rep. 1992, 10, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, Z.; Ren, W.; Han, F.; Yang, L.; Zhuang, M.; Lv, H.; Liu, Y.; Fang, Z.; Zhang, Y. Creation of fertility-restored materials for Ogura CMS in Brassica oleracea by introducing Rfo gene from Brassica napus via an allotriploid strategy. Theor. Appl. Genet. 2020, 133, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sharma, S.R.; Singh, B. Heterosis for mineral elements in single cross-hybrids of cabbage (Brassica oleracea var. capitata L.). Sci. Hortic. 2009, 122, 32–36. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, G.; Zhang, Y.; Song, Q.; Chen, Z.; Wang, J.; Guo, J.; Niu, N.; Wang, J.; Ma, S. Comparative studies of mitochondrial proteomics reveal an intimate protein network of male sterility in wheat (Triticum aestivum L.). J. Exp. Bot. 2015, 66, 6191–6203. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.L.H. Male Sterility in Higher Plants; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Wise, R.P.; Pring, D.R. Nuclear-mediated mitochondrial gene regulation and male fertility in higher plants: Light at the end of the tunnel? Proc. Natl. Acad. Sci. USA 2002, 99, 10240–10242. [Google Scholar] [CrossRef] [Green Version]
- Levings, C.S., 3rd; Pring, D.R. Restriction endonuclease analysis of mitochondrial DNA from normal and Texas cytoplasmic male-sterile maize. Science 1976, 193, 158–160. [Google Scholar] [CrossRef]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yoon, M.K. Comparison of mitochondrial and chloroplast genome segments from three onion (Allium cepa L.) cytoplasm types and identification of a trans-splicing intron of cox2. Curr. Genet. 2010, 56, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Dewey, R.E.; Timothy, D.H.; Levings, C.S. A mitochondrial protein associated with cytoplasmic male sterility in the T cytoplasm of maize. Proc. Natl. Acad. Sci. USA 1987, 84, 5374–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Gao, F.; Ji, Y.; Liu, Y.; Dan, Z.; Yang, P.; Zhu, Y.; Li, S. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef]
- Köhler, R.H.; Horn, R.; Lössl, A.; Zetsche, K. Cytoplasmic male sterility in sunflower is correlated with the co-transcription of a new open reading frame with the atpA gene. Mol. Gen. Genet. 1991, 227, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Huang, W.; Yin, C.; Gong, Z. Mitochondrial cytochrome c oxidase and F1Fo-ATPase dysfunction in peppers (Capsicum annuum L.) with cytoplasmic male sterility and its association with orf507 and Ψatp6-2 genes. Int. J. Mol. Sci. 2013, 14, 1050–1068. [Google Scholar] [CrossRef] [Green Version]
- Menassa, R.; L’Homme, Y.; Brown, G.G. Post-transcriptional and developmental regulation of a CMS-associated mitochondrial gene region by a nuclear restorer gene. Plant J. 1999, 17, 491–499. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuda, M.; Yasumoto, K.; Yamagishi, H.; Terachi, T. A complete mitochondrial genome sequence of Ogura-type male-sterile cytoplasm and its comparative analysis with that of normal cytoplasm in radish (Raphanus sativus L.). BMC Genom. 2012, 13, 352. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Kazama, T.; Murata, H.; Motomura, K.; Toriyama, K. Whole mitochondrial genome sequencing and transcriptional analysis to uncover an RT102-type cytoplasmic male sterility-associated candidate gene derived from Oryza rufipogon. Plant Cell Physiol. 2013, 54, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Hao, M.; Yang, W.; Li, T.; Shoaib, M.; Sun, J.; Liu, D.; Li, X.; Nie, Y.; Tian, X.; Zhang, A. Combined transcriptome and proteome analysis of anthers of AL-type cytoplasmic male sterile line and its maintainer line reveals new insights into mechanism of male sterility in common wheat. Front. Genet. 2021, 12, 762332. [Google Scholar] [CrossRef]
- Ye, J.; Duan, Y.; Hu, G.; Geng, X.; Zhang, G.; Yan, P.; Liu, Z.; Zhang, L.; Song, X. Identification of candidate genes and biosynthesis pathways related to fertility conversion by wheat KTM3315A transcriptome profiling. Front. Plant Sci. 2017, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bao, S.; Zhou, X.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Song, C.; Gao, L.; Zhang, H.; Yang, C.; Sheng, J.; Ren, J.; Chen, D.; Wang, Y. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in onion. Plant Physiol. Biochem. 2018, 125, 35–44. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Yang, Z.; Yi, B.; Wen, J.; Shen, J.; Tu, J.; Ma, C.; Fu, T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genom. 2014, 15, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Liu, Y.; Sun, Y.; Yang, A.; Li, F. Comparative transcriptome analysis reveals the potential mechanism of abortion in tobacco sua-cytoplasmic male sterility. Int. J. Mol. Sci. 2020, 21, 2445. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Wang, H.; Li, D.; Lin, Z.; Li, Y.; Zhao, W.; Chao, H.; Miao, L.; Li, M. Transcriptomic and proteomic analysis of Shaan2A cytoplasmic male sterility and its maintainer line in Brassica napus. Front. Plant Sci. 2019, 10, 252. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Tian, H.; Wang, S.; Shao, J.; Zheng, Y.; Zhang, H.; Guo, L.; Ding, Y. Pollen developmental defects in ZD-CMS rice line explored by cytological, molecular and proteomic approaches. J. Proteom. 2014, 108, 110–123. [Google Scholar] [CrossRef]
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Hu, K.; Yan, M.; Song, L.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Yi, B.; Tu, J. Mitochondrial genome and transcriptome analysis of five alloplasmic male-sterile lines in Brassica juncea. BMC Genom. 2019, 20, 348. [Google Scholar] [CrossRef]
- Ravet, K.; Pilon, M. Copper and iron homeostasis in plants: The challenges of oxidative stress. Antioxid. Redox Signal. 2013, 19, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Wu, S.; Li, Z.; Dong, Z.; An, X.; Ma, B.; Tian, Y.; Li, J. Maize genic male-sterility genes and their applications in hybrid breeding: Progress and perspectives. Mol. Plant 2019, 12, 321–342. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteom. 2018, 179, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, X.F.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; An, X.; Li, Z.; Yan, T.; Zhu, T.; Xie, K.; Liu, S.; Hou, Q.; Zhao, L.; Wu, S.; et al. CRISPR/Cas9-based discovery of maize transcription factors regulating male sterility and their functional conservation in plants. Plant Biotechnol. J. 2021, 19, 1769–1784. [Google Scholar] [CrossRef]
- Liu, Z.; Li, S.; Li, W.; Liu, Q.; Zhang, L.; Song, X. Comparative transcriptome analysis indicates that a core transcriptional network mediates isonuclear alloplasmic male sterility in wheat (Triticum aestivum L.). BMC Plant Biol. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackmore, S.; Wortley, A.H.; Skvarla, J.J.; Rowley, J.R. Pollen wall development in flowering plants. New Phytol. 2007, 174, 483–498. [Google Scholar] [CrossRef]
- Bu, Y.; Niu, F.; He, M.; Ye, J.; Yang, X.; Du, Z.; Zhang, L.; Song, X. The gene TaPG encoding a polygalacturonase is critical for pollen development and male fertility in thermo-sensitive cytoplasmic male-sterility wheat. Gene 2022, 833, 146596. [Google Scholar] [CrossRef]
- Pinzón-Latorre, D.; Deyholos, M.K. Characterization and transcript profiling of the pectin methylesterase (PME) and pectin methylesterase inhibitor (PMEI) gene families in flax (Linum usitatissimum). BMC Genom. 2013, 14, 742. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, L.; Wang, Y.; Wang, Y.C.; Wang, N.N.; Lu, R.; Wu, Y.W.; Li, X.B. Pollen-specific protein PSP231 activates callose synthesis to govern male gametogenesis and pollen germination. Plant Physiol. 2020, 184, 1024–1041. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Z.; Vizcay-Barrena, G.; Shi, J.; Liang, W.; Yuan, Z.; Werck-Reichhart, D.; Schreiber, L.; Wilson, Z.A.; Zhang, D. ABORTED MICROSPORES acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell. 2014, 26, 1544–1556. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, A.C.; Pearce, S.; Band, L.R.; Yang, C.; Ferjentsikova, I.; King, J.; Yuan, Z.; Zhang, D.; Wilson, Z.A. Biphasic regulation of the transcription factor ABORTED MICROSPORES (AMS) is essential for tapetum and pollen development in Arabidopsis. New Phytol. 2017, 213, 778–790. [Google Scholar] [CrossRef]
- Zhu, E.; You, C.; Wang, S.; Cui, J.; Niu, B.; Wang, Y.; Qi, J.; Ma, H.; Chang, F. The DYT1-interacting proteins bHLH010, bHLH089 and bHLH091 are redundantly required for Arabidopsis anther development and transcriptome. Plant J. 2015, 83, 976–990. [Google Scholar] [CrossRef]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and regulation of mitochondrial respiration in plants. Annu. Rev. Plant Biol. 2011, 62, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.M.; Subbaiah, C.C. Mitochondrial retrograde regulation in plants. Mitochondrion 2007, 7, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ma, N.; Wang, P.Y.; Fu, N.; Shen, H.L. Transcriptome sequencing and de novo analysis of a cytoplasmic male sterile line and its near-isogenic restorer line in chili pepper (Capsicum annuum L.). PLoS ONE 2013, 8, e65209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zancani, M.; Braidot, E.; Filippi, A.; Lippe, G. Structural and functional properties of plant mitochondrial F-ATP synthase. Mitochondrion 2020, 53, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Budar, F.; Pelletier, G. Male sterility in plants: Occurrence, determinism, significance and use. Comptes Rendus Acad. Sci. III. 2001, 324, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Geisler, D.A.; Päpke, C.; Obata, T.; Nunes-Nesi, A.; Matthes, A.; Schneitz, K.; Maximova, E.; Araújo, W.L.; Fernie, A.R.; Persson, S. Downregulation of the δ-subunit reduces mitochondrial ATP synthase levels, alters respiration, and restricts growth and gametophyte development in Arabidopsis. Plant Cell 2012, 24, 2792–2811. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pandeya, D.; Jo, Y.D.; Liu, W.Y.; Kang, B.C. Reduced activity of ATP synthase in mitochondria causes cytoplasmic male sterility in chili pepper. Planta 2013, 237, 1097–1109. [Google Scholar] [CrossRef]
- Yang, H.; Xue, Y.; Li, B.; Lin, Y.; Li, H.; Guo, Z.; Li, W.; Fu, Z.; Ding, D.; Tang, J. The chimeric gene atp6c confers cytoplasmic male sterility in maize by impairing the assembly of the mitochondrial ATP synthase complex. Mol. Plant. 2022, 15, 872–886. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, X.; Tan, Y.; Huang, J.B.; Zheng, Z.; Tao, L.Z. Phosphoethanolamine N-methyltransferase 1 contributes to maintenance of root apical meristem by affecting ROS and auxin-regulated cell differentiation in Arabidopsis. New Phytol. 2019, 224, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.; Lourenço, B.; Marques, M.; Ramalho-Santos, J. Mitochondria functionality and sperm quality. Reproduction 2013, 146, R163–R174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Shi, X.; Li, S.; Zhang, L.; Song, X. Oxidative stress and aberrant programmed cell death are associated with pollen abortion in isonuclear alloplasmic male-sterile wheat. Front. Plant Sci. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Shi, X.; Li, S.; Hu, G.; Zhang, L.; Song, X. Tapetal-delayed programmed cell death (PCD) and oxidative stress-induced male sterility of Aegilops uniaristata cytoplasm in wheat. Int. J. Mol. Sci. 2018, 19, 1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ren, W.; Zhang, B.; Chen, W.; Fang, Z.; Yang, L.; Zhuang, M.; Lv, H.; Wang, Y.; Ji, J.; et al. Organelle comparative genome analysis reveals novel alloplasmic male sterility with orf112 in Brassica oleracea L. Int. J. Mol. Sci. 2021, 22, 13230. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 19–616 | 19–2167 | FR2202 | |
|---|---|---|---|
| BaseNum (Gb) | 5.28 | 7.05 | 4.90 |
| ReadNum | 7,558,047 | 10,658,677 | 7,245,445 |
| PassReads | 7,557,374 | 10,657,713 | 7,244,891 |
| Primers_found | 6,429,226 | 9,214,366 | 6,313,210 |
| N50 | 897 | 857 | 841 |
| MeanLength | 697.7 | 656.3 | 680.3 |
| MaxLength | 183,372 | 128,123 | 113,851 |
| MeanQscore | 10.3 | 10.4 | 10.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Ren, W.; Zhang, B.; Guo, H.; Fang, Z.; Yang, L.; Zhuang, M.; Lv, H.; Wang, Y.; Ji, J.; et al. Comparative Transcriptome Analysis Reveals a Potential Regulatory Network for Ogura Cytoplasmic Male Sterility in Cabbage (Brassica oleracea L.). Int. J. Mol. Sci. 2023, 24, 6703. https://doi.org/10.3390/ijms24076703
Chen L, Ren W, Zhang B, Guo H, Fang Z, Yang L, Zhuang M, Lv H, Wang Y, Ji J, et al. Comparative Transcriptome Analysis Reveals a Potential Regulatory Network for Ogura Cytoplasmic Male Sterility in Cabbage (Brassica oleracea L.). International Journal of Molecular Sciences. 2023; 24(7):6703. https://doi.org/10.3390/ijms24076703
Chicago/Turabian StyleChen, Li, Wenjing Ren, Bin Zhang, Huiling Guo, Zhiyuan Fang, Limei Yang, Mu Zhuang, Honghao Lv, Yong Wang, Jialei Ji, and et al. 2023. "Comparative Transcriptome Analysis Reveals a Potential Regulatory Network for Ogura Cytoplasmic Male Sterility in Cabbage (Brassica oleracea L.)" International Journal of Molecular Sciences 24, no. 7: 6703. https://doi.org/10.3390/ijms24076703