
HED, a Human-Engineered Domain, Confers a Unique Fc-Binding Activity to Produce a New Class of Humanized Antibody-like Molecules
 and
and
Abstract
:1. Introduction
2. Results
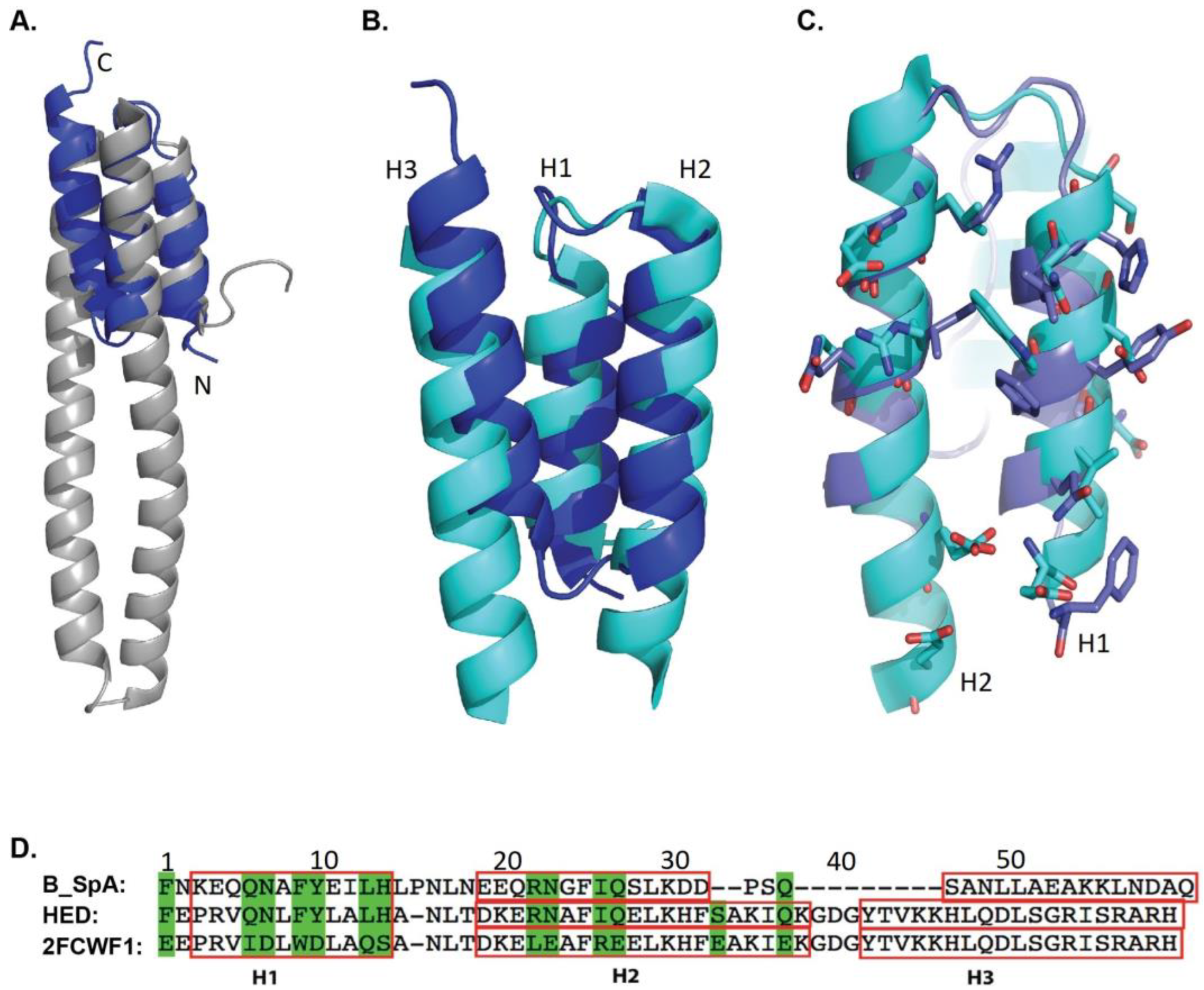
2.1. Model Design of HED
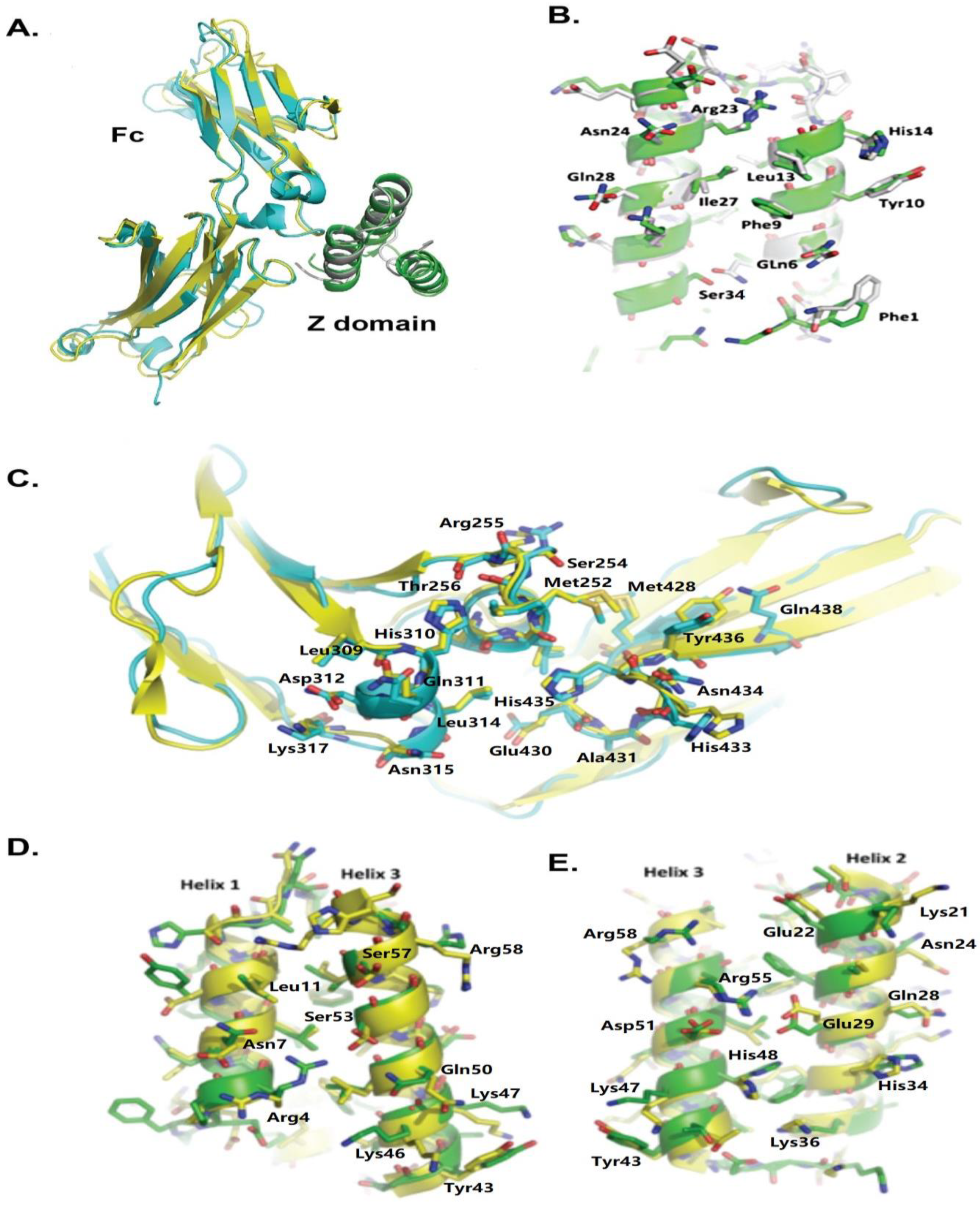
2.2. Crystal Structure of HED Complexed with hIgG1Fc
2.3. IgG Fc Associated with HED Binds to the Fc Receptor on Effector Cells
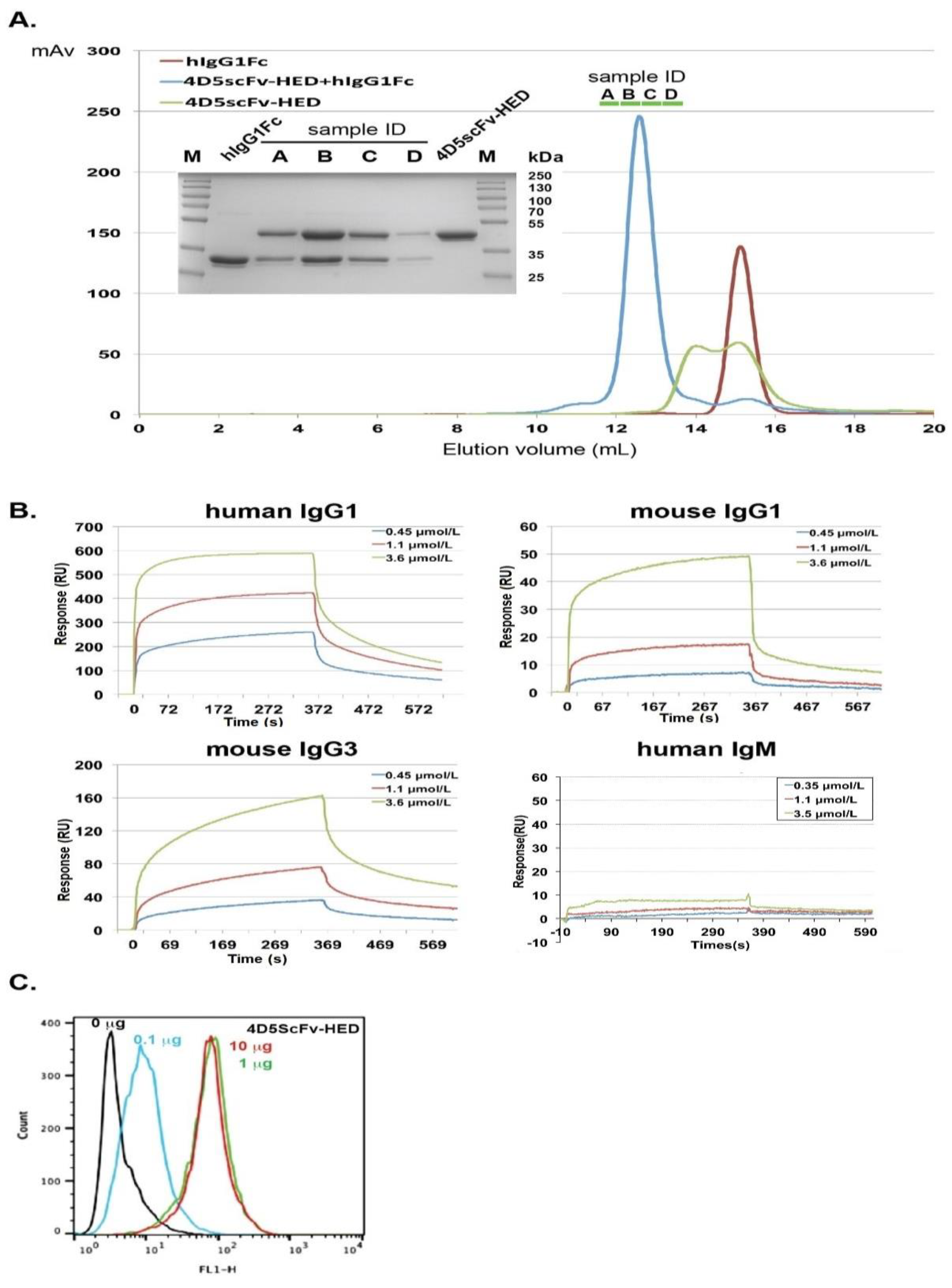
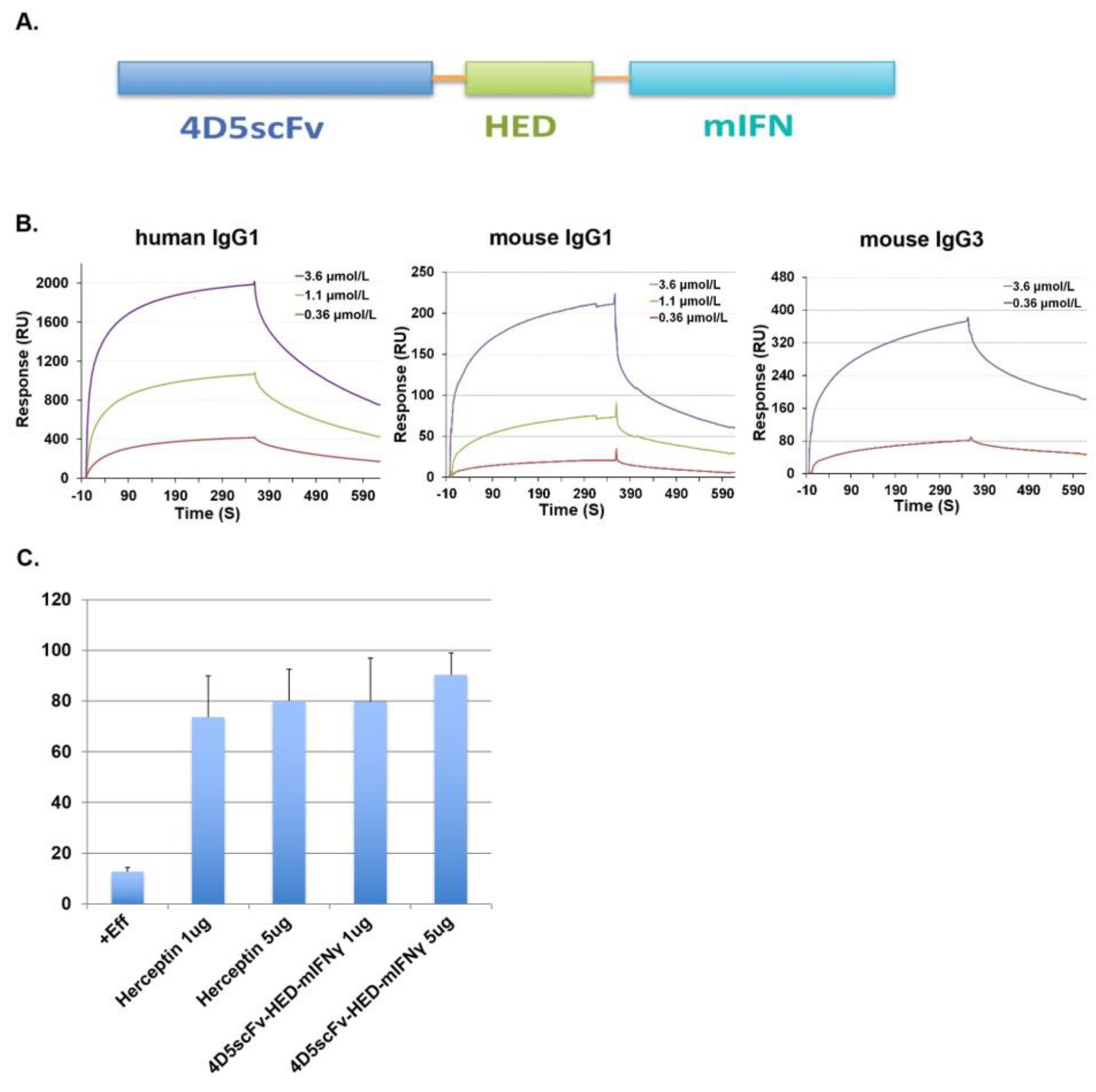
2.4. Fusion of scFv Fragments with HED to Produce an Antibody-like Structure That Potentiates ADCC and CDC by Recruiting hIgG1
2.5. Binding Potential of 4D5scFv-HED to Cell Surface p185Her2/neu
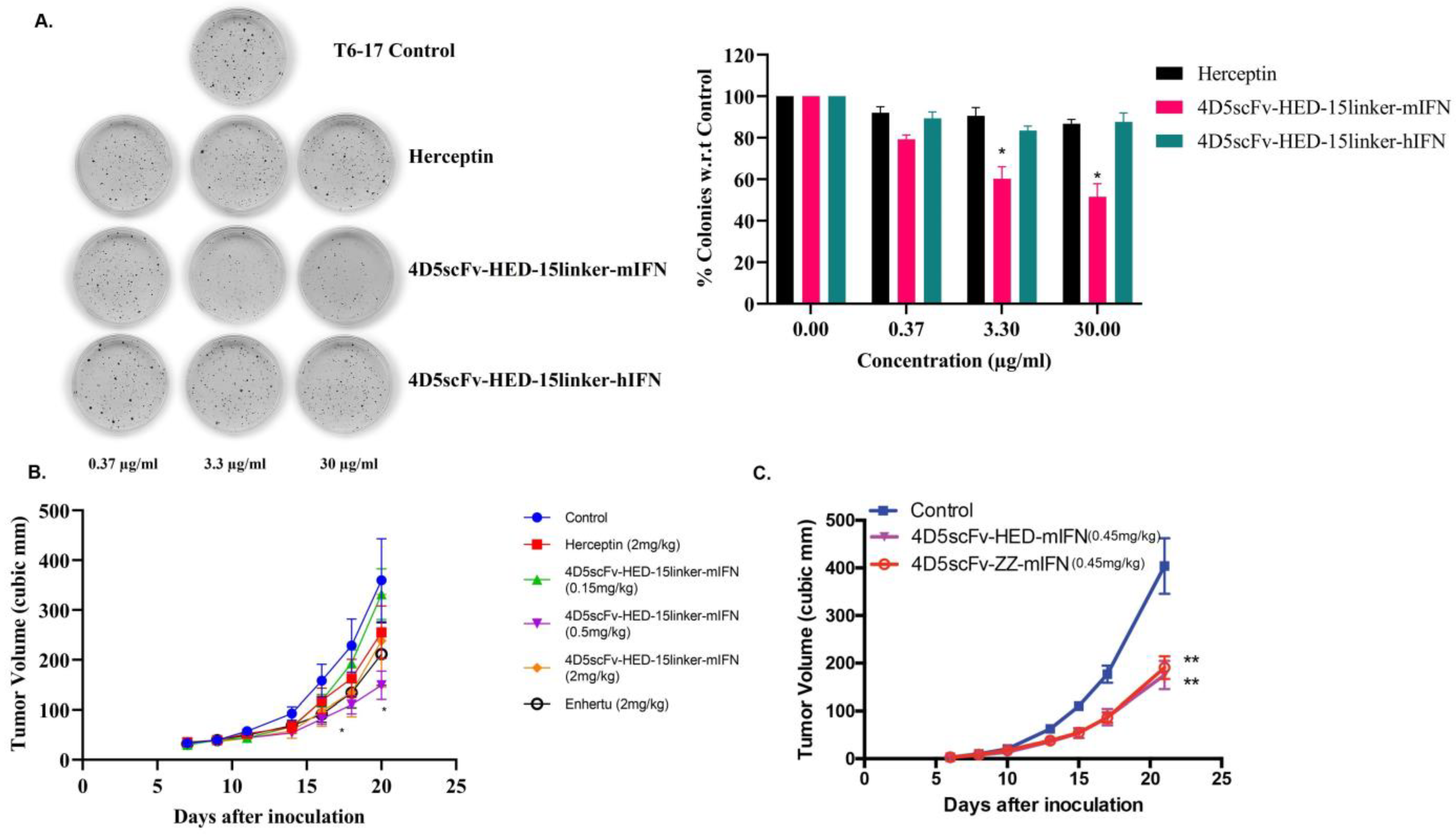
2.6. New Species of HED Bodies Inhibits Tumor Cell Growth In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Model Design
4.2. Protein Expression and Purification
4.3. Crystallization, Data Collection, and Structure Determination
4.4. Surface Plasmon Resonance (Biacore) Studies and FACS
4.5. ADCC Assay
4.6. Soft Agar Assay
4.7. In Vivo Xenograft Model
4.8. Study Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gomez, M.I.; O’Seaghdha, M.; Magargee, M.; Foster, T.J.; Prince, A.S. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J. Biol. Chem. 2006, 281, 20190–20196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartleib, J.; Kohler, N.; Dickinson, R.B.; Chhatwal, G.S.; Sixma, J.J.; Hartford, O.M.; Foster, T.J.; Peters, G.; Kehrel, B.E.; Herrmann, M. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 2000, 96, 2149–2156. [Google Scholar] [PubMed]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef]
- Hillson, J.L.; Karr, N.S.; Oppliger, I.R.; Mannik, M.; Sasso, E.H. The structural basis of germline-encoded VH3 immunoglobulin binding to staphylococcal protein A. J. Exp. Med. 1993, 178, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsgren, A.; Quie, P.G. Effects of staphylococcal protein A on heat labile opsonins. J. Immunol. 1974, 112, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, C.S.; Silverman, G.J. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. J. Exp. Med. 2003, 197, 1125–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graille, M.; Stura, E.A.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404. [Google Scholar] [CrossRef] [Green Version]
- Deis, L.N.; Pemble, C.W.t.; Qi, Y.; Hagarman, A.; Richardson, D.C.; Richardson, J.S.; Oas, T.G. Multiscale conformational heterogeneity in staphylococcal protein a: Possible determinant of functional plasticity. Structure 2014, 22, 1467–1477. [Google Scholar] [CrossRef] [Green Version]
- Stahl, S.; Nygren, P.A.; Uhlen, M. Detection and isolation of recombinant proteins based on binding affinity of reporter: Protein A. Methods Mol. Biol. 1997, 63, 103–118. [Google Scholar] [CrossRef]
- Cai, Z.; Fu, T.; Nagai, Y.; Lam, L.; Yee, M.; Zhu, Z.; Zhang, H. scFv-based “Grababody” as a general strategy to improve recruitment of immune effector cells to antibody-targeted tumors. Cancer Res. 2013, 73, 2619–2627. [Google Scholar] [CrossRef] [Green Version]
- Leonetti, M.; Thai, R.; Cotton, J.; Leroy, S.; Drevet, P.; Ducancel, F.; Boulain, J.C.; Menez, A. Increasing immunogenicity of antigens fused to Ig-binding proteins by cell surface targeting. J. Immunol. 1998, 160, 3820–3827. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Cheng, A.G.; Kim, H.Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Emolo, C.; DeDent, A.C.; Falugi, F.; Missiakas, D.M.; Schneewind, O. Protein A-specific monoclonal antibodies and prevention of Staphylococcus aureus disease in mice. Infect. Immun. 2012, 80, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Gouda, H.; Torigoe, H.; Saito, A.; Sato, M.; Arata, Y.; Shimada, I. Three-dimensional solution structure of the B domain of staphylococcal protein A: Comparisons of the solution and crystal structures. Biochemistry 1992, 31, 9665–9672. [Google Scholar] [CrossRef]
- Sato, S.; Religa, T.L.; Daggett, V.; Fersht, A.R. Testing protein-folding simulations by experiment: B domain of protein A. Proc. Natl. Acad. Sci. USA 2004, 101, 6952–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Fisher, C.; Beglova, N.; Blacklow, S.C. Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol. Cell 2006, 22, 277–283. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Kesmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothe, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [Green Version]
- Deis, L.N.; Wu, Q.; Wang, Y.; Qi, Y.; Daniels, K.G.; Zhou, P.; Oas, T.G. Suppression of conformational heterogeneity at a protein-protein interface. Proc. Natl. Acad. Sci. USA 2015, 112, 9028–9033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Ramsland, P.A.; Farrugia, W.; Bradford, T.M.; Sardjono, C.T.; Esparon, S.; Trist, H.M.; Powell, M.S.; Tan, P.S.; Cendron, A.C.; Wines, B.D.; et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J. Immunol. 2011, 187, 3208–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Braisted, A.C.; Wells, J.A. Minimizing a binding domain from protein A. Proc. Natl. Acad. Sci. USA 1996, 93, 5688–5692. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Tsuchiya, H.; Runkle, E.A.; Young, P.D.; Ji, M.Q.; Norton, L.; Drebin, J.A.; Zhang, H.; Greene, M.I. Disabling of the erbB Pathway Followed by IFN-gamma Modifies Phenotype and Enhances Genotoxic Eradication of Breast Tumors. Cell Rep. 2015, 12, 2049–2059. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lam, L.; Nagai, Y.; Zhu, Z.; Chen, X.; Ji, M.Q.; Greene, M.I. A targeted immunotherapy approach for HER2/neu transformed tumors by coupling an engineered effector domain with interferon-γ. OncoImmunology 2018, 7, e1300739. [Google Scholar] [CrossRef] [Green Version]
- Masuda, K.; Richter, M.; Song, X.; Berezov, A.; Murali, R.; Greene, M.I.; Zhang, H. AHNP-streptavidin: A tetrameric bacterially produced antibody surrogate fusion protein against p185her2/neu. Oncogene 2006, 25, 7740–7746. [Google Scholar] [CrossRef] [Green Version]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Delivery of molecular medicine to solid tumors: Lessons from in vivo imaging of gene expression and function. J. Control. Release 2001, 74, 7–25. [Google Scholar] [CrossRef]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Sanz, L.; Cuesta, A.M.; Compte, M.; Alvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef]
- Sethu, S.; Govindappa, K.; Alhaidari, M.; Pirmohamed, M.; Park, K.; Sathish, J. Immunogenicity to biologics: Mechanisms, prediction and reduction. Arch. Immunol. Ther. Exp. (Warsz) 2012, 60, 331–344. [Google Scholar] [CrossRef]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Cytokine resurrection: Engineered IL-2 ramps up immuno-oncology responses. Nat. Biotechnol. 2018, 36, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Ou, S.S.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P.; et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J. Clin. Oncol. 2008, 26, 5422–5428. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Hsu, E.; Fu, Y.X.; Peng, H. Next-generation cytokines for cancer immunotherapy. Antib. Ther. 2021, 4, 123–133. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values |
|---|---|
| Data Collection | |
| Space group | P31 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 100.16, 100.16, 84.57 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 20–2.7 (2.75–2.70) a |
| Rmerge | 0.072 (0.956) |
| Average I/σI | 52.2 (3.0) |
| Completeness (%) | 99.9 (100) |
| Structure Refinement | |
| Resolution (Å) | 20–2.70 |
| Unique reflections | 13,703 |
| Unique reflections for Rfree | 724 |
| No. of protein atoms/solvent | 2199/31 |
| Rcrystallography (%) | 19.13 (30.5) |
| Rfree (%) | 24.25 (43.0) |
| R.M.S deviations | |
| Bond length (Å)/angle (°) | 0.008/1.207 |
| Average B factor (Å2) | 83.50 |
| Ramachandran plot | |
| Most favored (%) | 93 |
| Allowed (%) | 6.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Goel, P.N.; Zheng, C.; Nagai, Y.; Lam, L.; Samanta, A.; Ji, M.; Zhang, H.; Greene, M.I. HED, a Human-Engineered Domain, Confers a Unique Fc-Binding Activity to Produce a New Class of Humanized Antibody-like Molecules. Int. J. Mol. Sci. 2023, 24, 6477. https://doi.org/10.3390/ijms24076477
Zhu Z, Goel PN, Zheng C, Nagai Y, Lam L, Samanta A, Ji M, Zhang H, Greene MI. HED, a Human-Engineered Domain, Confers a Unique Fc-Binding Activity to Produce a New Class of Humanized Antibody-like Molecules. International Journal of Molecular Sciences. 2023; 24(7):6477. https://doi.org/10.3390/ijms24076477
Chicago/Turabian StyleZhu, Zhiqiang, Peeyush N. Goel, Cai Zheng, Yasuhiro Nagai, Lian Lam, Arabinda Samanta, Meiqing Ji, Hongtao Zhang, and Mark I. Greene. 2023. "HED, a Human-Engineered Domain, Confers a Unique Fc-Binding Activity to Produce a New Class of Humanized Antibody-like Molecules" International Journal of Molecular Sciences 24, no. 7: 6477. https://doi.org/10.3390/ijms24076477