
Inflammation and Organ Injury the Role of Substance P and Its Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Substance P and Its Receptors
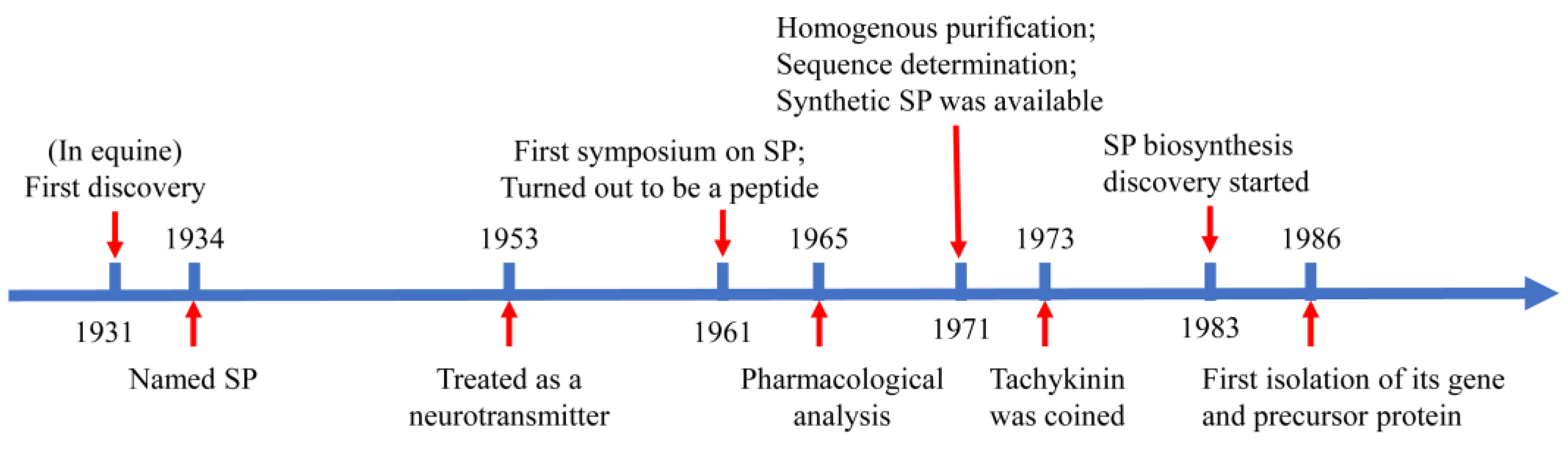
2.1. Overview of Substance P
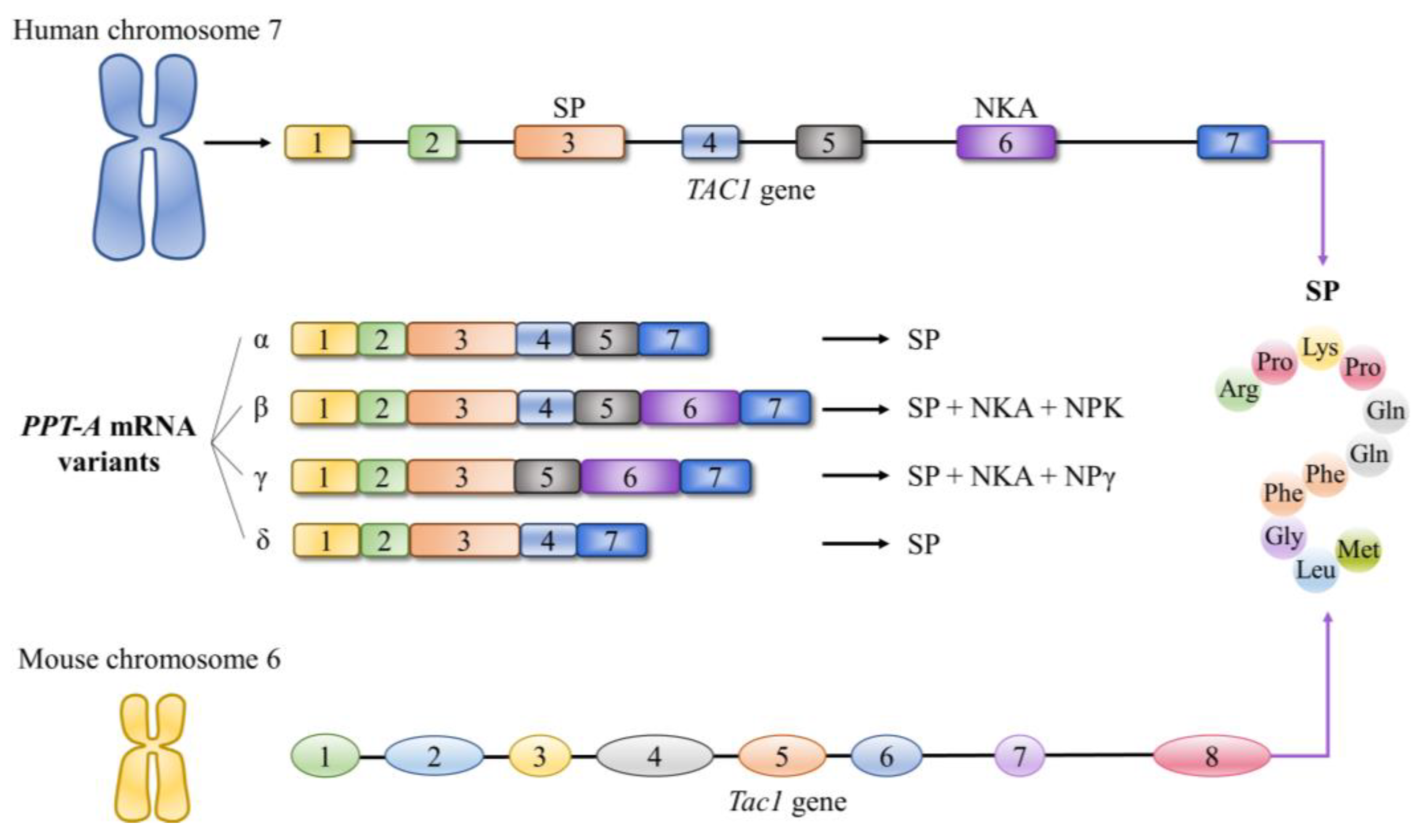
2.2. Overview of Substance P Receptors
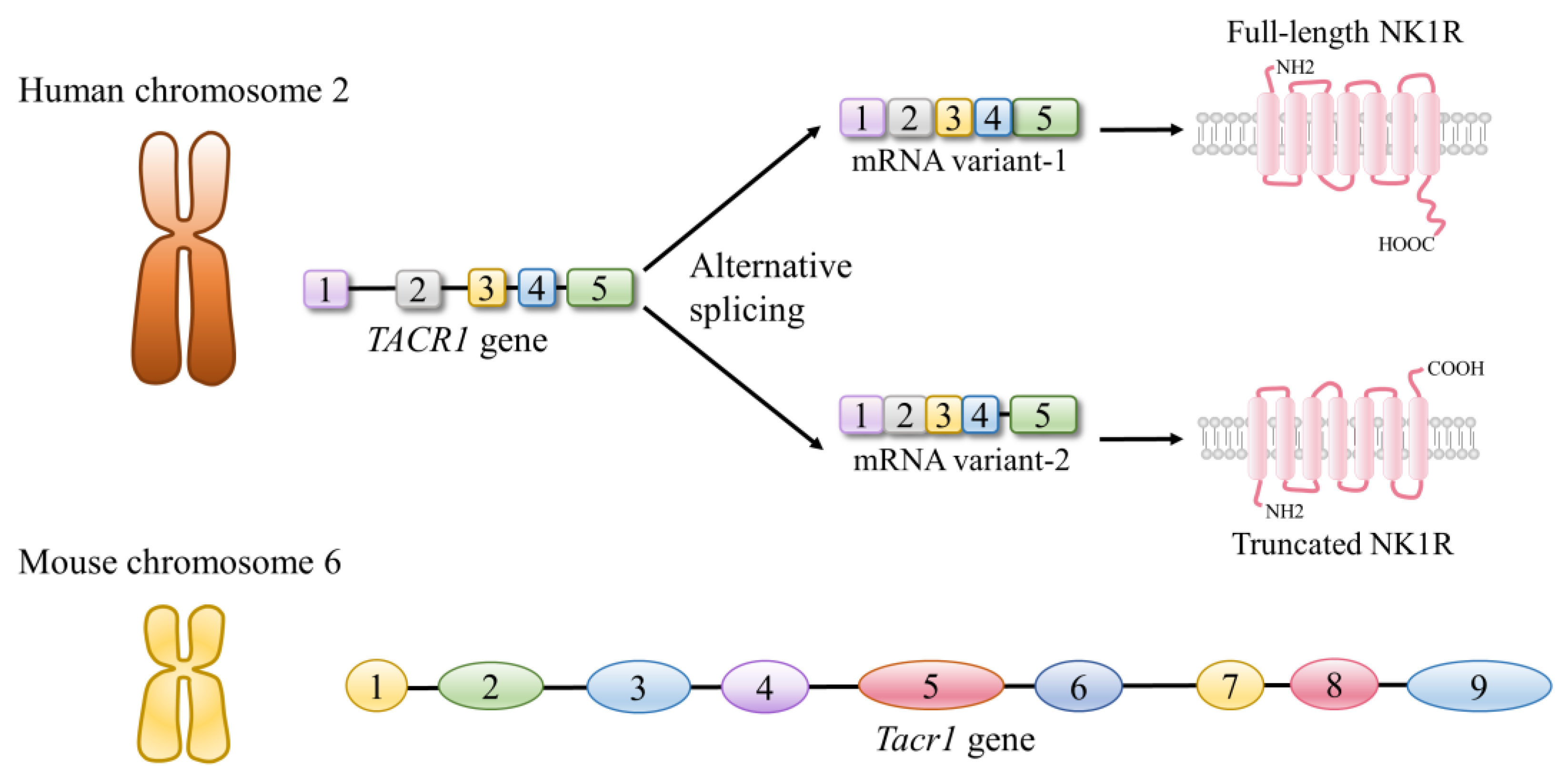
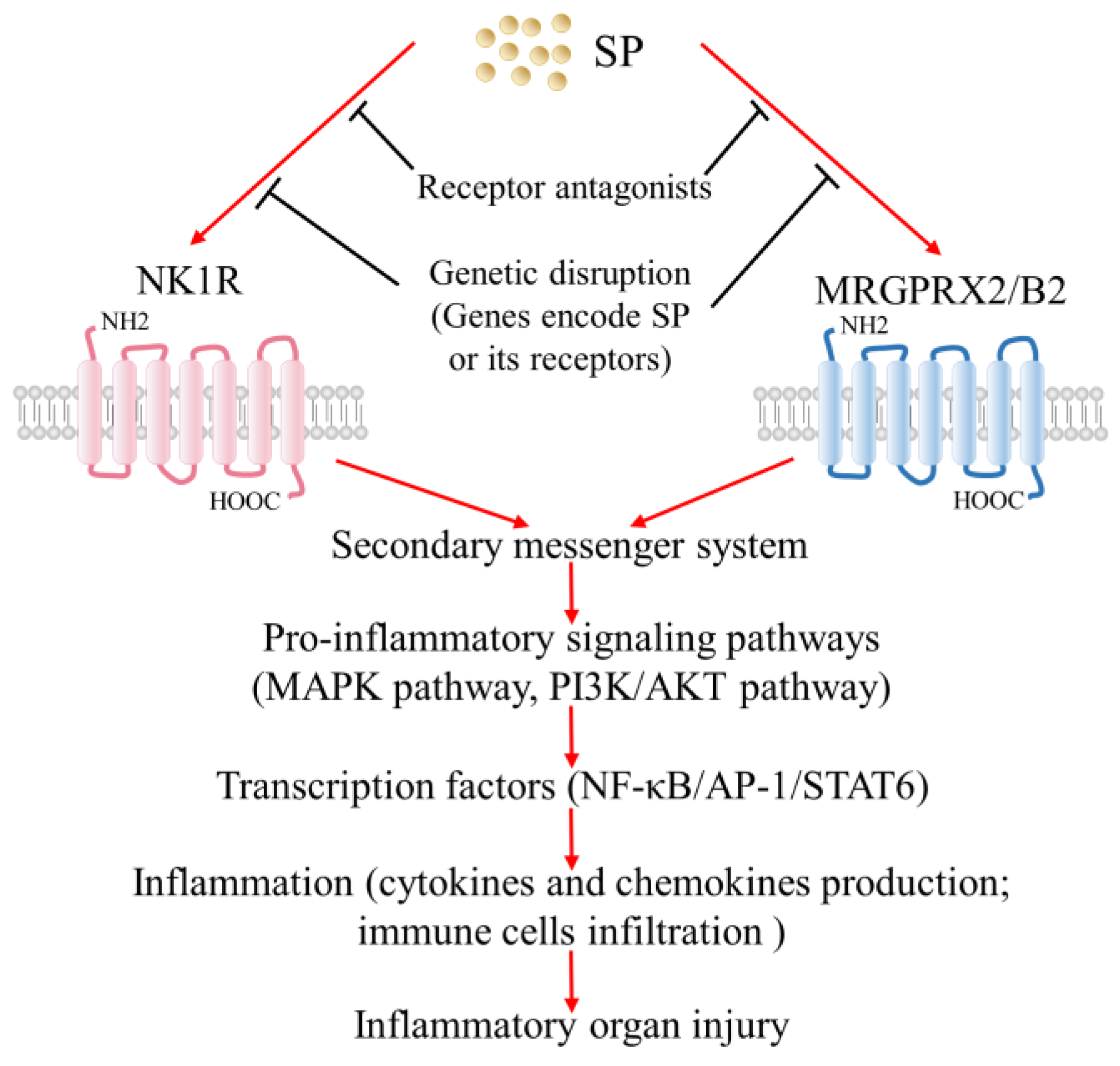
2.2.1. Neurokinin Receptors
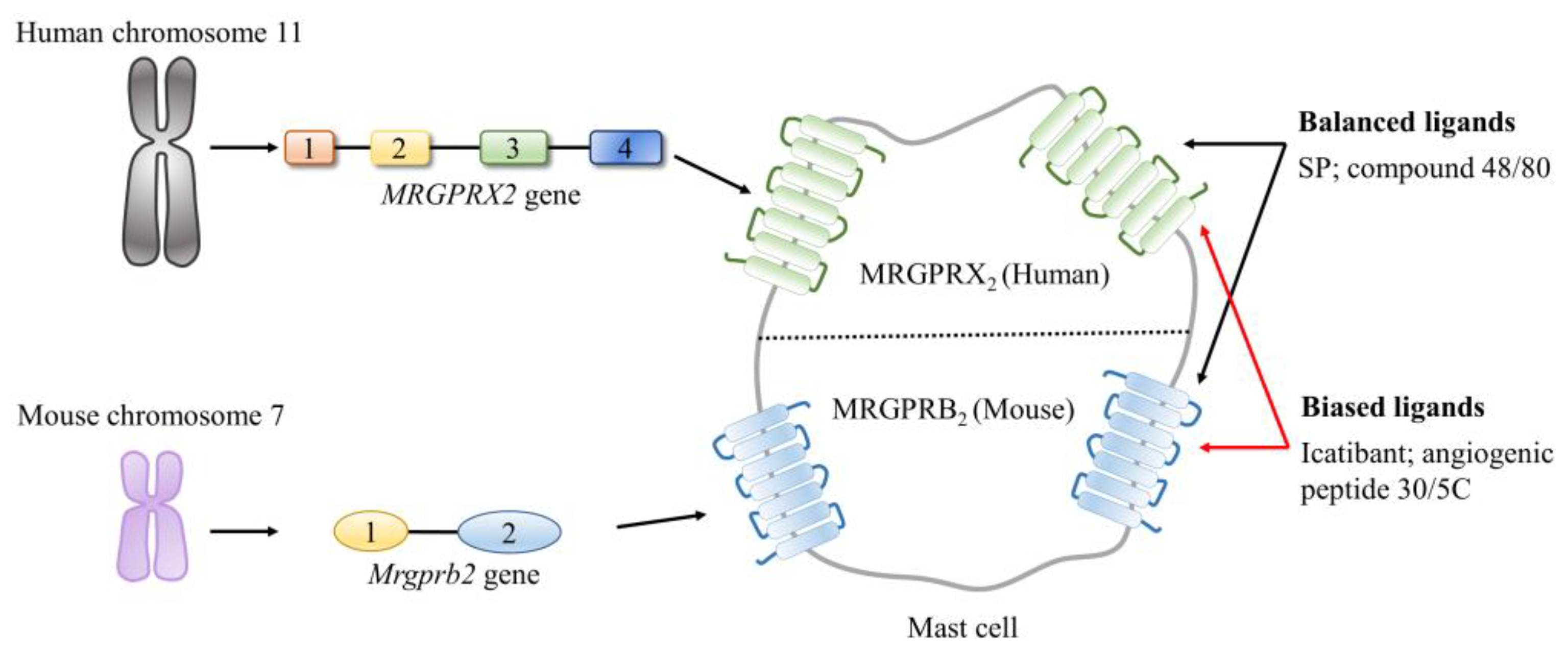
2.2.2. Mas-Related G Protein-Coupled Receptors (MRGPRs)
3. Substance P and Its Receptors in Inflammation-Associated Organ Injury
3.1. Emerging Roles of the SP–NK1R System in Inflammation-Associated Organ Injury
3.1.1. Role of the SP–NK1R System in Sepsis-Related Multiple Organ Injury
3.1.2. Roles of the SP–NK1R System in Acute Pancreatitis-Related Lung Injury
3.1.3. Roles of the SP–NK1R System in Burn Injury Associated Lung Injury
3.2. Emerging Roles of the SP–MRGPRX2/B2 System in Inflammation-Associated Organ Injury
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oronsky, B.; Caroen, S.; Reid, T. What Exactly Is Inflammation (and What Is It Not?). Int. J. Mol. Sci. 2022, 23, 14905. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C. Nonresolving inflammation redux. Immunity 2022, 55, 592–605. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef]
- Zhu, C.J.; Yang, W.G.; Li, D.J.; Song, Y.D.; Chen, S.Y.; Wang, Q.F.; Liu, Y.N.; Zhang, Y.; Cheng, B.; Wu, Z.W.; et al. Calycosin attenuates severe acute pancreatitis-associated acute lung injury by curtailing high mobility group box 1—Induced inflammation. World J. Gastroenterol. 2021, 27, 7669–7686. [Google Scholar] [CrossRef]
- Comish, P.B.; Liu, M.M.; Huebinger, R.; Carlson, D.; Kang, R.; Tang, D. The cGAS-STING pathway connects mitochondrial damage to inflammation in burn-induced acute lung injury in rat. Burns 2022, 48, 168–175. [Google Scholar] [CrossRef]
- Song, Y.; Miao, S.; Li, Y.; Fu, H. Ulinastatin attenuates liver injury and inflammation in a cecal ligation and puncture induced sepsis mouse model. J. Cell Biochem. 2019, 120, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Choaib, A.; Issa, E.; El Choueiry, F.; Eldin, J.N.; Shbaklo, K.; Alhajj, M.; Sawaya, R.T.; Assi, G.; Nader, M.; Chatila, R.; et al. SARS-CoV-2-mediated liver injury: Pathophysiology and mechanisms of disease. Inflamm. Res. 2022, 72, 1–12. [Google Scholar] [CrossRef]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Zeng, Y.; Chen, X. Exosomes derived from adipose-derived stem cells alleviate cigarette smoke-induced lung inflammation and injury by inhibiting alveolar macrophages pyroptosis. Respir. Res. 2022, 23, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Cornide-Petronio, M.E.; Gracia-Sancho, J.; Peralta, C. Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells 2019, 8, 1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US, V.E.; Gaddum, J.H. An unidentified depressor substance in certain tissue extracts. J. Physiol. 1931, 72, 74–87. [Google Scholar] [CrossRef]
- Zieglgänsberger, W. Substance P and pain chronicity. Cell Tissue Res. 2019, 375, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Suvas, S. Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J. Immunol. 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [Green Version]
- Khorasani, S.; Boroumand, N.; Lavi Arab, F.; Hashemy, S.I. The immunomodulatory effects of tachykinins and their receptors. J. Cell Biochem. 2020, 121, 3031–3041. [Google Scholar] [CrossRef]
- Redkiewicz, P. The Regenerative Potential of Substance P. Int. J. Mol. Sci. 2022, 23, 750. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Alalikhan, A.; Aghaee-Bakhtiari, S.H.; Hashemy, S.I. The redox modulatory effects of SP/NK1R system: Implications for oxidative stress-associated disorders. Life Sci. 2022, 296, 120448. [Google Scholar] [CrossRef]
- Thapaliya, M.; Chompunud Na Ayudhya, C.; Amponnawarat, A.; Roy, S.; Ali, H. Mast Cell-Specific MRGPRX2: A Key Modulator of Neuro-Immune Interaction in Allergic Diseases. Curr. Allergy Asthma Rep. 2021, 21, 3. [Google Scholar] [CrossRef]
- Kumar, A.; Bhatia, M. Role of Hydrogen Sulfide, Substance P and Adhesion Molecules in Acute Pancreatitis. Int. J. Mol. Sci. 2021, 22, 12136. [Google Scholar] [CrossRef]
- Gaddam, R.R.; Chambers, S.; Murdoch, D.; Shaw, G.; Bhatia, M. Circulating levels of hydrogen sulfide and substance P in patients with sepsis. J. Infect. 2017, 75, 293–300. [Google Scholar] [CrossRef]
- Sio, S.W.; Ang, S.F.; Lu, J.; Moochhala, S.; Bhatia, M. Substance P upregulates cyclooxygenase-2 and prostaglandin E metabolite by activating ERK1/2 and NF-kappaB in a mouse model of burn-induced remote acute lung injury. J. Immunol. 2010, 185, 6265–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, K.R.; Lee, H.; Han, S.H.; Ahn, W.; Kim, D.K.; Kim, I.S.; Jung, B.S.; Lee, S. Substance P, A Promising Therapeutic Target in Musculoskeletal Disorders. Int. J. Mol. Sci. 2022, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Coveñas, R.; Muñoz, M. Involvement of the Substance P/Neurokinin-1 Receptor System in Cancer. Cancers 2022, 14, 3539. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Yosipovitch, G. Substance P and neurokinin 1 receptor are new targets for the treatment of chronic pruritus. Br. J. Dermatol. 2019, 181, 932–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nat. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [Green Version]
- Pennefather, J.N.; Lecci, A.; Candenas, M.L.; Patak, E.; Pinto, F.M.; Maggi, C.A. Tachykinins and tachykinin receptors: A growing family. Life Sci. 2004, 74, 1445–1463. [Google Scholar] [CrossRef]
- Navratilova, E.; Porreca, F. Substance P and Inflammatory Pain: Getting It Wrong and Right Simultaneously. Neuron 2019, 101, 353–355. [Google Scholar] [CrossRef]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [Green Version]
- Kleczkowska, P.; Nowicka, K.; Bujalska-Zadrozny, M.; Hermans, E. Neurokinin-1 receptor-based bivalent drugs in pain management: The journey to nowhere? Pharmacol. Ther. 2019, 196, 44–58. [Google Scholar] [CrossRef]
- Wei, X.L.; Luo, L.; Chen, M.Z.; Zhou, J.; Lan, B.Y.; Ma, X.M.; Chen, W.X. Temporospatial Expression of Neuropeptide Substance P in Dental Pulp Stem Cells during Odontoblastic Differentiation in Vitro and Reparative Dentinogenesis in Vivo. J. Endod. 2022, 49, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Douglas, S.D.; Ho, W. Human stem cells express substance P gene and its receptor. J. Hematotherapy Stem Cell Res. 2000, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Valentin-Hansen, L.; Park, M.; Huber, T.; Grunbeck, A.; Naganathan, S.; Schwartz, T.W.; Sakmar, T.P. Mapping substance P binding sites on the neurokinin-1 receptor using genetic incorporation of a photoreactive amino acid. J. Biol. Chem. 2014, 289, 18045–18054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.A.; Faust, B.; Gondin, A.B.; Dämgen, M.A.; Suomivuori, C.M.; Veldhuis, N.A.; Cheng, Y.; Dror, R.O.; Thal, D.M.; Manglik, A. Selective G protein signaling driven by substance P-neurokinin receptor dynamics. Nat. Chem. Biol. 2022, 18, 109–115. [Google Scholar] [CrossRef]
- Saidi, M.; Kamali, S.; Beaudry, F. Characterization of Substance P processing in mouse spinal cord S9 fractions using high-resolution Quadrupole-Orbitrap mass spectrometry. Neuropeptides 2016, 59, 47–55. [Google Scholar] [CrossRef]
- Grady, E.F.; Garland, A.M.; Gamp, P.D.; Lovett, M.; Payan, D.G.; Bunnett, N.W. Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol. Biol. Cell 1995, 6, 509–524. [Google Scholar] [CrossRef] [Green Version]
- Sankhe, R.; Pai, S.R.K.; Kishore, A. Tumour suppression through modulation of neprilysin signaling: A comprehensive review. Eur. J. Pharmacol. 2021, 891, 173727. [Google Scholar] [CrossRef]
- Rameshwar, P.; Joshi, D.D.; Yadav, P.; Qian, J.; Gascon, P.; Chang, V.T.; Anjaria, D.; Harrison, J.S.; Song, X. Mimicry between neurokinin-1 and fibronectin may explain the transport and stability of increased substance P immunoreactivity in patients with bone marrow fibrosis. Blood 2001, 97, 3025–3031. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Chambers, S.; Zeng, Y.; Bhatia, M. Gases in Sepsis: Novel Mediators and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3669. [Google Scholar] [CrossRef]
- Li, Y.S.; Xi, Y.; Li, X.J.; Leng, C.L.; Jia, M.M.; Zhang, W.K.; Tang, H.B. Up-Regulation of the Biosynthesis and Release of Substance P through Wnt/β-Catenin Signaling Pathway in Rat Dorsal Root Ganglion Cells. PLoS ONE 2015, 10, e0129701. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Setiawan, T.; Hang, L.; Stoyanoff, K.; Weinstock, J.V. Interleukin-12 (IL-12) and IL-23 induction of substance p synthesis in murine T cells and macrophages is subject to IL-10 and transforming growth factor beta regulation. Infect. Immun. 2008, 76, 3651–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schank, J.R.; Heilig, M. Substance P and the Neurokinin-1 Receptor: The New CRF. Int. Rev. Neurobiol. 2017, 136, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Green, D.P.; Limjunyawong, N.; Gour, N.; Pundir, P.; Dong, X. A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron 2019, 101, 412–420.e3. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskaya, K.; Hostettler, A.; Buergi, S.; Draeger, A. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J. Biol. Chem. 2005, 280, 7135–7146. [Google Scholar] [CrossRef] [Green Version]
- Thom, C.; Ehrenmann, J.; Vacca, S.; Waltenspühl, Y.; Schöppe, J.; Medalia, O.; Plückthun, A. Structures of neurokinin 1 receptor in complex with G(q) and G(s) proteins reveal substance P binding mode and unique activation features. Sci. Adv. 2021, 7, eabk2872. [Google Scholar] [CrossRef]
- Cottrell, G.S.; Padilla, B.; Pikios, S.; Roosterman, D.; Steinhoff, M.; Gehringer, D.; Grady, E.F.; Bunnett, N.W. Ubiquitin-dependent down-regulation of the neurokinin-1 receptor. J. Biol. Chem. 2006, 281, 27773–27783. [Google Scholar] [CrossRef] [Green Version]
- Spitsin, S.; Pappa, V.; Douglas, S.D. Truncation of neurokinin-1 receptor-Negative regulation of substance P signaling. J. Leukoc. Biol. 2018, 103, 1043–1051. [Google Scholar] [CrossRef]
- Lai, J.P.; Ho, W.Z.; Kilpatrick, L.E.; Wang, X.; Tuluc, F.; Korchak, H.M.; Douglas, S.D. Full-length and truncated neurokinin-1 receptor expression and function during monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 7771–7776. [Google Scholar] [CrossRef] [Green Version]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar]
- Caberlotto, L.; Hurd, Y.L.; Murdock, P.; Wahlin, J.P.; Melotto, S.; Corsi, M.; Carletti, R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur. J. Neurosci. 2003, 17, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 12605–12610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.T.; Jiang, B.Y.; Chen, C.C. Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. Int. J. Mol. Sci. 2019, 20, 1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PubChem, Explore Chemistry. Quickly Find Chemical Information from Authoritative Sources. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 15 March 2023).
- Meixiong, J.; Dong, X. Mas-Related G Protein-Coupled Receptors and the Biology of Itch Sensation. Annu. Rev. Genet. 2017, 51, 103–121. [Google Scholar] [CrossRef]
- Yang, F.; Guo, L.; Li, Y.; Wang, G.; Wang, J.; Zhang, C.; Fang, G.X.; Chen, X.; Liu, L.; Yan, X.; et al. Structure, function and pharmacology of human itch receptor complexes. Nature 2021, 600, 164–169. [Google Scholar] [CrossRef]
- Solinski, H.J.; Gudermann, T.; Breit, A. Pharmacology and signaling of MAS-related G protein-coupled receptors. Pharmacol. Rev. 2014, 66, 570–597. [Google Scholar] [CrossRef] [Green Version]
- Al Hamwi, G.; Riedel, Y.K.; Clemens, S.; Namasivayam, V.; Thimm, D.; Müller, C.E. MAS-related G protein-coupled receptors X (MRGPRX): Orphan GPCRs with potential as targets for future drugs. Pharmacol. Ther. 2022, 238, 108259. [Google Scholar] [CrossRef]
- Roy, S.; Chompunud Na Ayudhya, C.; Thapaliya, M.; Deepak, V.; Ali, H. Multifaceted MRGPRX2: New insight into the role of mast cells in health and disease. J. Allergy Clin. Immunol. 2021, 148, 293–308. [Google Scholar] [CrossRef]
- Quan, P.L.; Sabaté-Brescó, M.; Guo, Y.; Martín, M.; Gastaminza, G. The Multifaceted Mas-Related G Protein-Coupled Receptor Member X2 in Allergic Diseases and Beyond. Int. J. Mol. Sci. 2021, 22, 4421. [Google Scholar] [CrossRef]
- Wedi, B.; Gehring, M.; Kapp, A. The pseudoallergen receptor MRGPRX2 on peripheral blood basophils and eosinophils: Expression and function. Allergy 2020, 75, 2229–2242. [Google Scholar] [CrossRef] [Green Version]
- Kiatsurayanon, C.; Niyonsaba, F.; Chieosilapatham, P.; Okumura, K.; Ikeda, S.; Ogawa, H. Angiogenic peptide (AG)-30/5C activates human keratinocytes to produce cytokines/chemokines and to migrate and proliferate via MrgX receptors. J. Dermatol. Sci. 2016, 83, 190–199. [Google Scholar] [CrossRef]
- Serhan, N.; Cenac, N.; Basso, L.; Gaudenzio, N. Mas-related G protein-coupled receptors (Mrgprs)—Key regulators of neuroimmune interactions. Neurosci. Lett. 2021, 749, 135724. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, H.; Noguchi, M. Therapeutic Potential of MRGPRX2 Inhibitors on Mast Cells. Cells 2021, 10, 2906. [Google Scholar] [CrossRef] [PubMed]
- Chompunud Na Ayudhya, C.; Amponnawarat, A.; Ali, H. Substance P Serves as a Balanced Agonist for MRGPRX2 and a Single Tyrosine Residue Is Required for β-Arrestin Recruitment and Receptor Internalization. Int. J. Mol. Sci. 2021, 22, 5318. [Google Scholar] [CrossRef] [PubMed]
- Lazki-Hagenbach, P.; Kleeblatt, E.; Ali, H.; Sagi-Eisenberg, R. Spatiotemporal Patterns of Substance P-Bound MRGPRX2 Reveal a Novel Connection Between Macropinosome Resolution and Secretory Granule Regeneration in Mast Cells. Front. Immunol. 2022, 13, 892239. [Google Scholar] [CrossRef]
- Roy, S.; Ganguly, A.; Haque, M.; Ali, H. Angiogenic Host Defense Peptide AG-30/5C and Bradykinin B(2) Receptor Antagonist Icatibant Are G Protein Biased Agonists for MRGPRX2 in Mast Cells. J. Immunol. 2019, 202, 1229–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, D.; Kashiwakura, J.; Kita, H.; Kikukawa, Y.; Fujitani, Y.; Sasaki-Sakamoto, T.; Kuroda, K.; Nunomura, S.; Hayama, K.; Terui, T.; et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J. Allergy Clin. Immunol. 2014, 134, 622–633.e9. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. H₂S and substance P in inflammation. Methods Enzym. 2015, 555, 195–205. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Manandhar, S.; Sinha, P.; Ejiwale, G.; Bhatia, M. Hydrogen Sulfide and its Interaction with Other Players in Inflammation. Adv. Exp. Med. Biol. 2021, 1315, 129–159. [Google Scholar] [CrossRef]
- Puneet, P.; Hegde, A.; Ng, S.W.; Lau, H.Y.; Lu, J.; Moochhala, S.M.; Bhatia, M. Preprotachykinin-A gene products are key mediators of lung injury in polymicrobial sepsis. J. Immunol. 2006, 176, 3813–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.; Zhang, H.; Moochhala, S.M.; Bhatia, M. Neurokinin-1 receptor antagonist treatment protects mice against lung injury in polymicrobial sepsis. J. Leukoc. Biol. 2007, 82, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.; Koh, Y.H.; Moochhala, S.M.; Bhatia, M. Neurokinin-1 receptor antagonist treatment in polymicrobial sepsis: Molecular insights. Int. J. Inflam. 2010, 2010, 601098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.; Tamizhselvi, R.; Manikandan, J.; Melendez, A.J.; Moochhala, S.M.; Bhatia, M. Substance P in polymicrobial sepsis: Molecular fingerprint of lung injury in preprotachykinin-A−/− mice. Mol. Med. 2010, 16, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Mella, J.R.; Stucchi, A.F.; Duffy, E.R.; Remick, D.G. Neurokinin-1 Receptor Deficiency Improves Survival in Murine Polymicrobial Sepsis Through Multiple Mechanisms in Aged Mice. Shock 2019, 52, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Chambers, S.; Fraser, R.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis. Int. J. Mol. Sci. 2019, 20, 3191. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Zhang, H.; Hegde, A.; Bhatia, M. Role of preprotachykinin-A gene products on multiple organ injury in LPS-induced endotoxemia. J. Leukoc. Biol. 2008, 83, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhong, D.; Dong, P.; Song, Y. Blocking CXCR1/2 contributes to amelioration of lipopolysaccharide-induced sepsis by downregulating substance P. J. Cell Biochem. 2018, 120, 2007–2014. [Google Scholar] [CrossRef]
- Arnalich, F.; Hernanz, A.; Jiménez, M.; López, J.; Tato, E.; Vázquez, J.J.; Montiel, C. Relationship between circulating levels of calcitonin gene-related peptide, nitric oxide metabolites and hemodynamic changes in human septic shock. Regul. Pept. 1996, 65, 115–121. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Almeida, T.; Hernández, M.; Ferreres, J.; Solé-Violán, J.; Labarta, L.; Díaz, C.; Jiménez, A. Association between serum substance P levels and mortality in patients with severe sepsis. J. Crit. Care 2015, 30, 924–928. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; Ferreres, J.; Solé-Violán, J.; Labarta, L.; Díaz, C.; Jiménez, A. Sustained Low Serum Substance P Levels in Non-Surviving Septic Patients. Int. J. Mol. Sci. 2017, 18, 1531. [Google Scholar] [CrossRef] [Green Version]
- Verdrengh, M.; Tarkowski, A. The impact of substance P signalling on the development of experimental staphylococcal sepsis and arthritis. Scand. J. Immunol. 2008, 67, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.B. Acute Pancreatitis. Ann. Intern. Med. 2021, 174, Itc17–Itc32. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.Y.; Tan, M.L.; Wu, L.M.; Asrani, V.M.; Windsor, J.A.; Yadav, D.; Petrov, M.S. Global incidence and mortality of pancreatic diseases: A systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol. Hepatol. 2016, 1, 45–55. [Google Scholar] [CrossRef]
- Figini, M.; Emanueli, C.; Grady, E.F.; Kirkwood, K.; Payan, D.G.; Ansel, J.; Gerard, C.; Geppetti, P.; Bunnett, N. Substance P and bradykinin stimulate plasma extravasation in the mouse gastrointestinal tract and pancreas. Am. J. Physiol. 1997, 272, G785–G793. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Saluja, A.K.; Hofbauer, B.; Frossard, J.L.; Lee, H.S.; Castagliuolo, I.; Wang, C.C.; Gerard, N.; Pothoulakis, C.; Steer, M.L. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc. Natl. Acad. Sci. USA 1998, 95, 4760–4765. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, M.; Slavin, J.; Cao, Y.; Basbaum, A.I.; Neoptolemos, J.P. Preprotachykinin-A gene deletion protects mice against acute pancreatitis and associated lung injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G830–G836. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.Y.; Wong, F.L.; Bhatia, M. A key role of neurokinin 1 receptors in acute pancreatitis and associated lung injury. Biochem. Biophys. Res. Commun. 2005, 327, 509–515. [Google Scholar] [CrossRef]
- Koh, Y.H.; Moochhala, S.; Bhatia, M. The role of neutral endopeptidase in caerulein-induced acute pancreatitis. J. Immunol. 2011, 187, 5429–5439. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Bhatia, M. Blockade of neurokinin-1 receptor attenuates CC and CXC chemokine production in experimental acute pancreatitis and associated lung injury. Am. J. Physiology. Gastrointest. Liver Physiol. 2007, 292, G143–G153. [Google Scholar] [CrossRef] [Green Version]
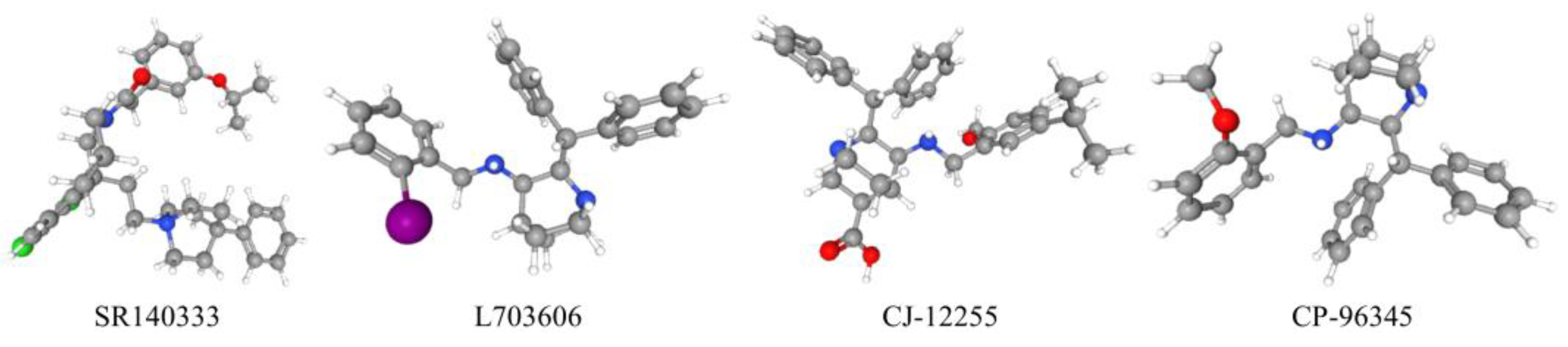
- Lau, H.Y.; Bhatia, M. Effect of CP-96,345 on the expression of adhesion molecules in acute pancreatitis in mice. Am. J. Physiology. Gastrointest. Liver Physiol. 2007, 292, G1283–G1292. [Google Scholar] [CrossRef] [Green Version]
- Ramnath, R.D.; Sun, J.; Bhatia, M. Involvement of SRC family kinases in substance P-induced chemokine production in mouse pancreatic acinar cells and its significance in acute pancreatitis. J. Pharmacol. Exp. Ther. 2009, 329, 418–428. [Google Scholar] [CrossRef]
- Li, B.; Han, X.; Ye, X.; Ni, J.; Wu, J.; Dai, J.; Wu, Z.; Chen, C.; Wan, R.; Wang, X.; et al. Substance P-regulated leukotriene B4 production promotes acute pancreatitis-associated lung injury through neutrophil reverse migration. Int. Immunopharmacol. 2018, 57, 147–156. [Google Scholar] [CrossRef]
- Amiti; Tamizhselvi, R.; Manickam, V. Menadione (vitamin K3) inhibits hydrogen sulfide and substance P via NF-κB pathway in caerulein-induced acute pancreatitis and associated lung injury in mice. Pancreatology 2019, 19, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Du, D.; Wen, Y.; Li, J.; Wang, R.; Jin, T.; Yang, J.; Shi, N.; Jiang, K.; Deng, L.; et al. Chaiqin chengqi decoction ameliorates acute pancreatitis in mice via inhibition of neuron activation-mediated acinar cell SP/NK1R signaling pathways. J. Ethnopharmacol. 2021, 274, 114029. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; van Baar, M.E.; Choudhry, M.A.; Chung, K.K.; Gibran, N.S.; Logsetty, S. Burn injury. Nat. Rev. Dis. Prim. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zou, B.; Liou, Y.C.; Huang, C. The pathogenesis and diagnosis of sepsis post burn injury. Burn. Trauma 2021, 9, tkaa047. [Google Scholar] [CrossRef] [PubMed]
- Knuth, C.M.; Auger, C.; Jeschke, M.G. Burn-induced hypermetabolism and skeletal muscle dysfunction. Am. J. Physiol. Cell Physiol. 2021, 321, C58–C71. [Google Scholar] [CrossRef]
- Xie, C.; Hu, J.; Cheng, Y.; Yao, Z. Researches on cognitive sequelae of burn injury: Current status and advances. Front. Neurosci. 2022, 16, 1026152. [Google Scholar] [CrossRef]
- Sio, S.W.; Puthia, M.K.; Lu, J.; Moochhala, S.; Bhatia, M. The neuropeptide substance P is a critical mediator of burn-induced acute lung injury. J. Immunol. 2008, 180, 8333–8341. [Google Scholar] [CrossRef] [Green Version]
- Sio, S.W.; Moochhala, S.; Lu, J.; Bhatia, M. Early protection from burn-induced acute lung injury by deletion of preprotachykinin-A gene. Am. J. Respir. Crit. Care Med. 2010, 181, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Deyo, D.J.; Cox, R.A.; Jacob, R.K.; Herndon, D.N.; Traber, D.L.; Hawkins, H.K. Substance P antagonist CP-96345 blocks lung vascular leakage and inflammation more effectively than its stereoisomer CP-96344 in a mouse model of smoke inhalation and burn injury. Toxicol. Mech. Methods 2010, 20, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, H.; Furuno, M.; Edamura, K.; Noguchi, M. Novel MRGPRX2 antagonists inhibit IgE-independent activation of human umbilical cord blood-derived mast cells. J. Leukoc. Biol. 2019, 106, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Chaki, S.; Alkanfari, I.; Roy, S.; Amponnawarat, A.; Hui, Y.; Oskeritzian, C.A.; Ali, H. Inhibition of Orai Channel Function Regulates Mas-Related G Protein-Coupled Receptor-Mediated Responses in Mast Cells. Front. Immunol. 2021, 12, 803335. [Google Scholar] [CrossRef] [PubMed]
- Che, D.; Zheng, Y.; Hou, Y.; Du, X.; Jia, T.; Zhao, Q.; Song, X.; Zhou, T.; Geng, S. Action of substance P and PAMP(9–20) on different excitation sites of MRGPRX2 induces differences in mast cell activation. Int. Immunopharmacol. 2021, 101, 108342. [Google Scholar] [CrossRef] [PubMed]
- Hsin, L.; Fernandopulle, N.A.; Ding, J.; Lumb, C.; Veldhuis, N.; Karas, J.A.; Northfield, S.E.; Mackay, G.A. The effect of substance P and its common in vivo-formed metabolites on MRGPRX2 and human mast cell activation. Pharmacol. Res. Perspect 2022, 10, e00990. [Google Scholar] [CrossRef]
- Antia, C.; Baquerizo, K.; Korman, A.; Bernstein, J.A.; Alikhan, A. Urticaria: A comprehensive review: Epidemiology, diagnosis, and work-up. J. Am. Acad. Dermatol. 2018, 79, 599–614. [Google Scholar] [CrossRef]
- Gonçalo, M.; Gimenéz-Arnau, A.; Al-Ahmad, M.; Ben-Shoshan, M.; Bernstein, J.A.; Ensina, L.F.; Fomina, D.; Galvàn, C.A.; Godse, K.; Grattan, C.; et al. The global burden of chronic urticaria for the patient and society. Br. J. Dermatol. 2021, 184, 226–236. [Google Scholar] [CrossRef]
- Lang, D.M. Chronic Urticaria. N. Engl. J. Med. 2022, 387, 824–831. [Google Scholar] [CrossRef]
- Church, M.K.; Kolkhir, P.; Metz, M.; Maurer, M. The role and relevance of mast cells in urticaria. Immunol. Rev. 2018, 282, 232–247. [Google Scholar] [CrossRef]
- Vena, G.A.; Cassano, N.; Di Leo, E.; Calogiuri, G.F.; Nettis, E. Focus on the role of substance P in chronic urticaria. Clin. Mol. Allergy 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, M.; Krull, C.; Hawro, T.; Saluja, R.; Groffik, A.; Stanger, C.; Staubach, P.; Maurer, M. Substance P is upregulated in the serum of patients with chronic spontaneous urticaria. J. Investig. Dermatol. 2014, 134, 2833–2836. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Wang, J.; Zhu, W.; Xu, C.; He, S. Upregulated expression of substance P in basophils of the patients with chronic spontaneous urticaria: Induction of histamine release and basophil accumulation by substance P. Cell Biol. Toxicol. 2016, 32, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Fadaee, J.; Khoshkhui, M.; Emadzadeh, M.; Hashemy, S.I.; Farid Hosseini, R.; Jabbari Azad, F.; Ahanchian, H.; Lavi Arab, F. Evaluation of Serum Substance P Level in Chronic Urticaria and Correlation with Disease Severity. Iran. J. Allergy Asthma Immunol. 2020, 19, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Yumoto, K.; Kawakami, T. An improved mouse model of atopic dermatitis and suppression of skin lesions by an inhibitor of Tec family kinases. Allergol. Int. 2007, 56, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, N.; Basso, L.; Sibilano, R.; Petitfils, C.; Meixiong, J.; Bonnart, C.; Reber, L.L.; Marichal, T.; Starkl, P.; Cenac, N.; et al. House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat. Immunol. 2019, 20, 1435–1443. [Google Scholar] [CrossRef]
- Wang, N.; Wang, J.; Zhang, Y.; Zeng, Y.; Hu, S.; Bai, H.; Hou, Y.; Wang, C.; He, H.; He, L. Imperatorin ameliorates mast cell-mediated allergic airway inflammation by inhibiting MRGPRX2 and CamKII/ERK signaling pathway. Biochem. Pharmacol. 2021, 184, 114401. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Y.; Zeng, Y.; Hu, S.; Bai, H.; Wang, J.; Jing, H.; Wang, N. Licochalcone A inhibits MAS-related GPR family member X2-induced pseudo-allergic reaction by suppressing nuclear migration of nuclear factor-κB. Phytother. Res. 2021, 35, 6270–6280. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Bhatia, M. Inflammation and Organ Injury the Role of Substance P and Its Receptors. Int. J. Mol. Sci. 2023, 24, 6140. https://doi.org/10.3390/ijms24076140
Zhu Z, Bhatia M. Inflammation and Organ Injury the Role of Substance P and Its Receptors. International Journal of Molecular Sciences. 2023; 24(7):6140. https://doi.org/10.3390/ijms24076140
Chicago/Turabian StyleZhu, Zhixing, and Madhav Bhatia. 2023. "Inflammation and Organ Injury the Role of Substance P and Its Receptors" International Journal of Molecular Sciences 24, no. 7: 6140. https://doi.org/10.3390/ijms24076140