
PD-1 Independent Role of PD-L1 in Triple-Negative Breast Cancer Progression
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
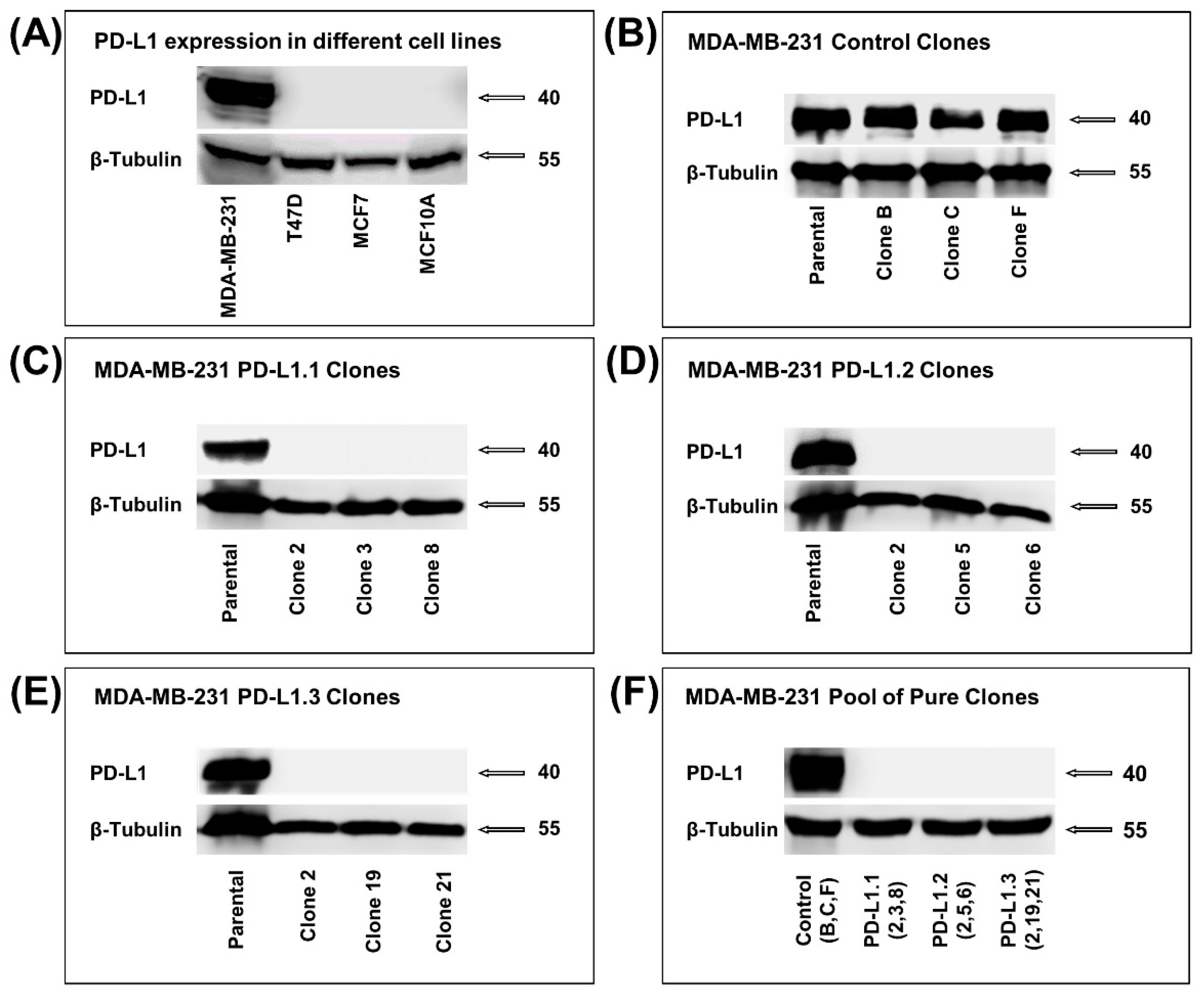
2.1. PD-L1 Knockout in TNBC MDA-MB-231 Cell Line
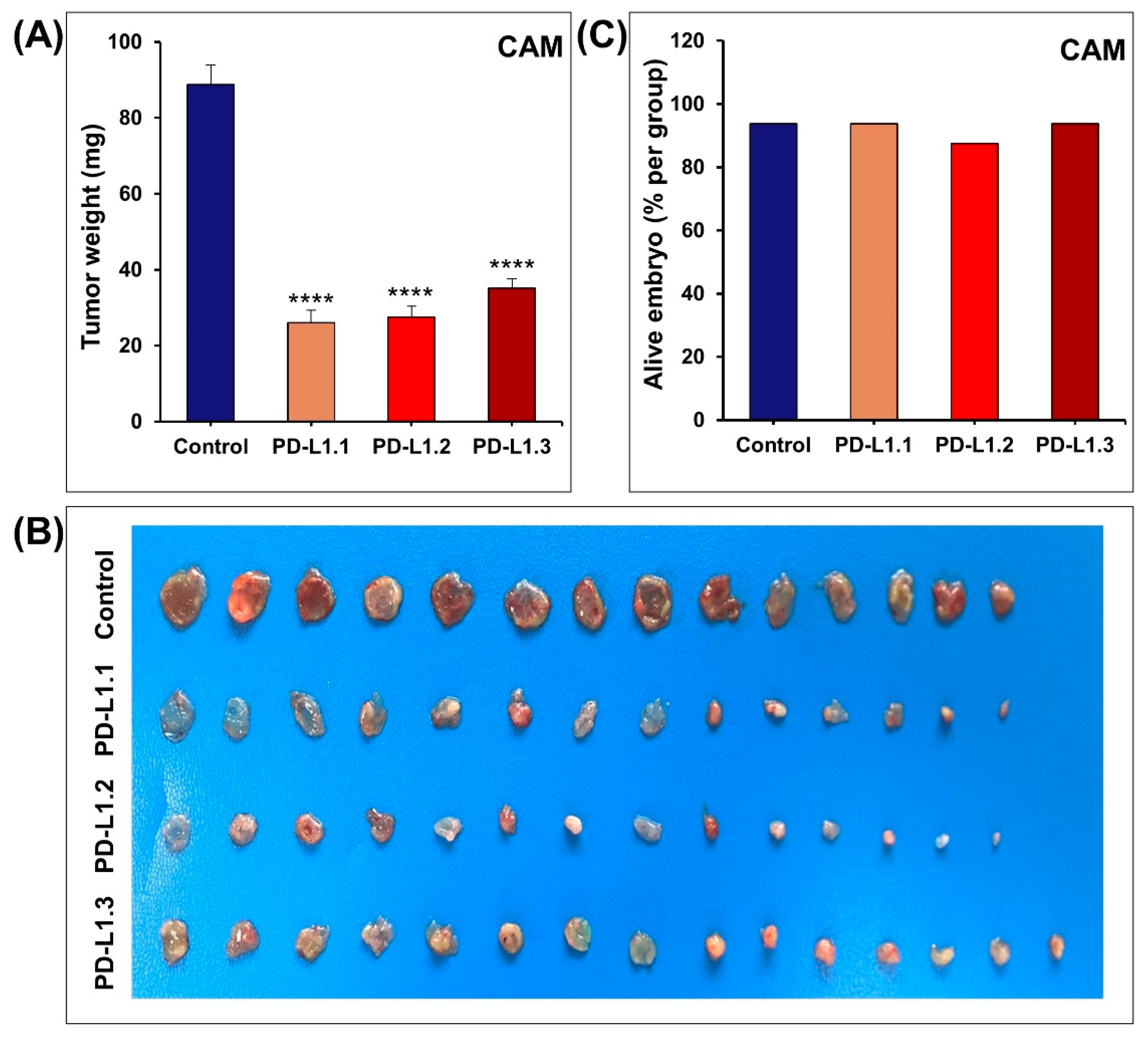
2.2. PD-L1 Knockout Decreases MDA-MB-231 Cells Proliferation, Colony Formation In Vitro, and Tumor Growth In Vivo
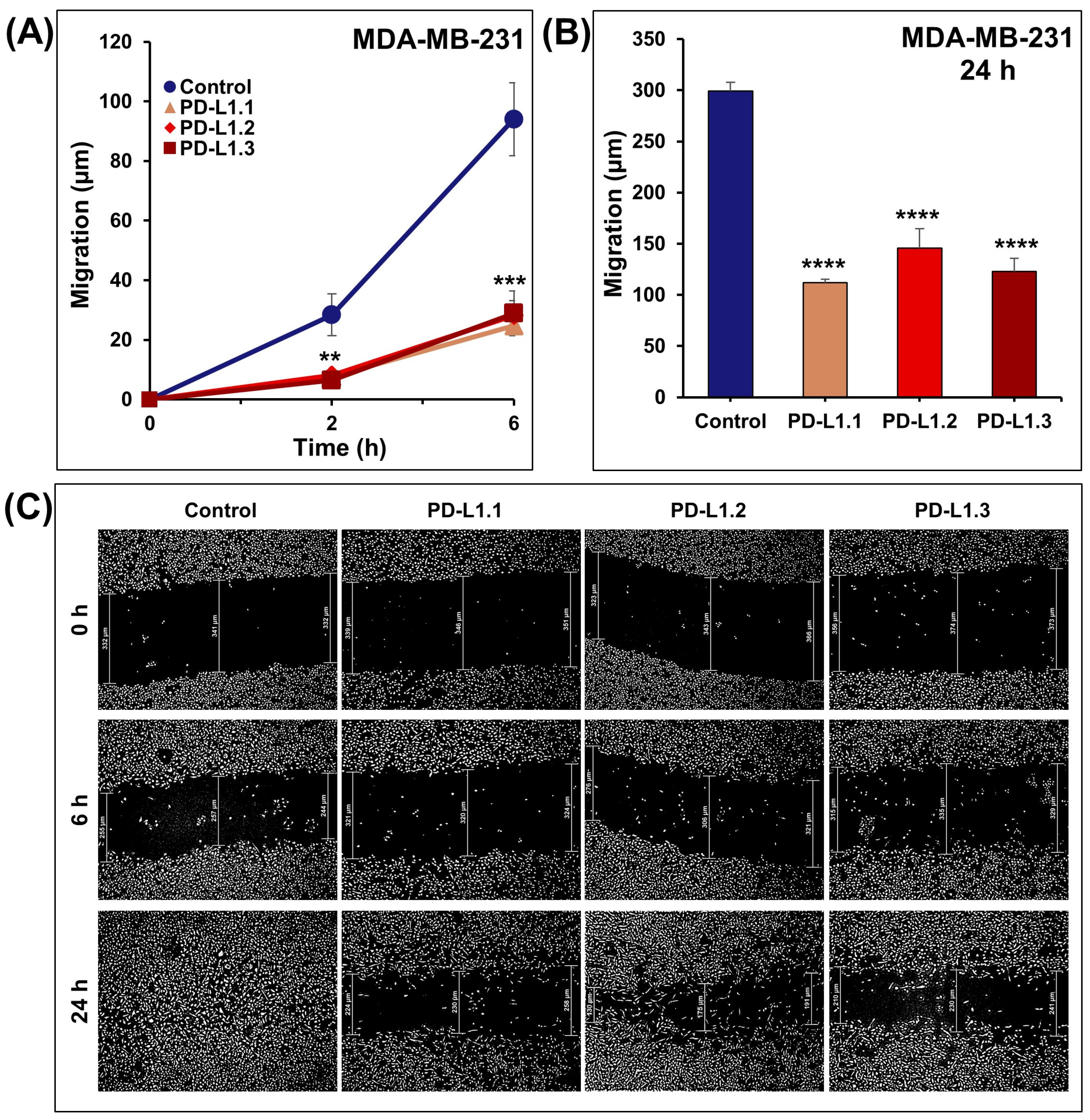
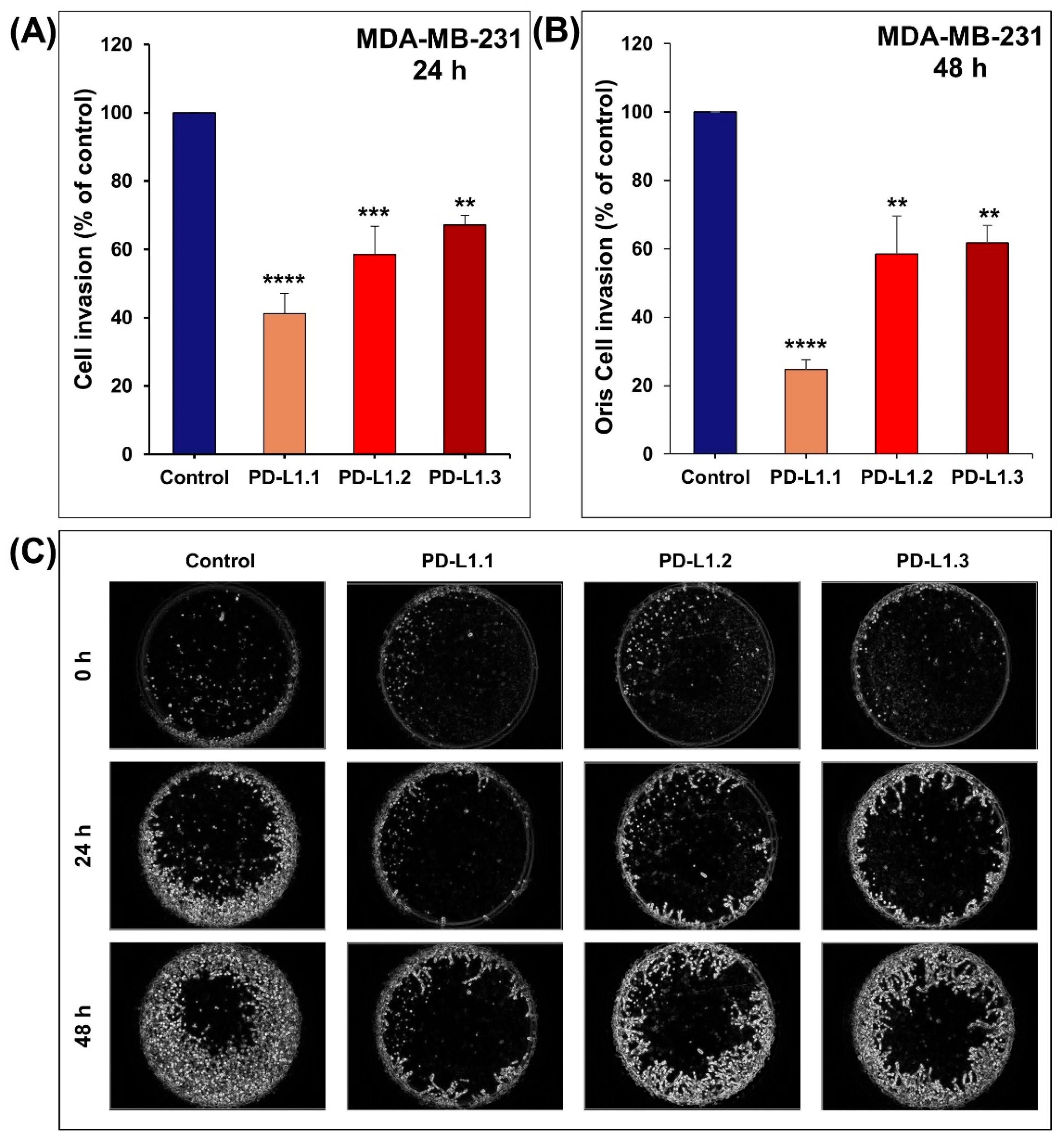
2.3. PD-L1 Knockout Decreases MDA-MB-231 Cell Migration and Invasion In Vitro
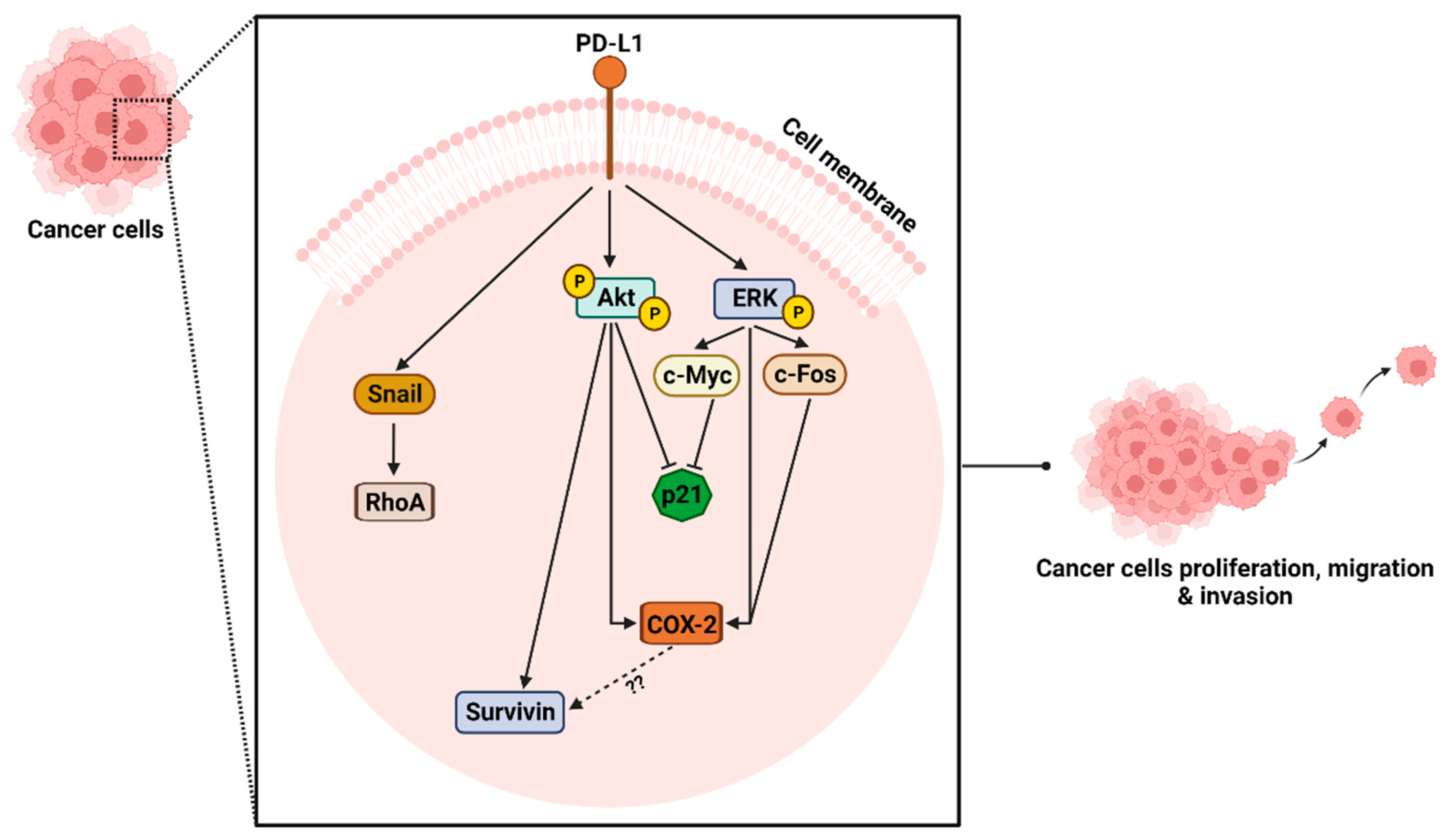
2.4. The Role of Signaling Pathways in PD-L1 Mediated Proliferation, Migration, Invasion, and Tumor Growth
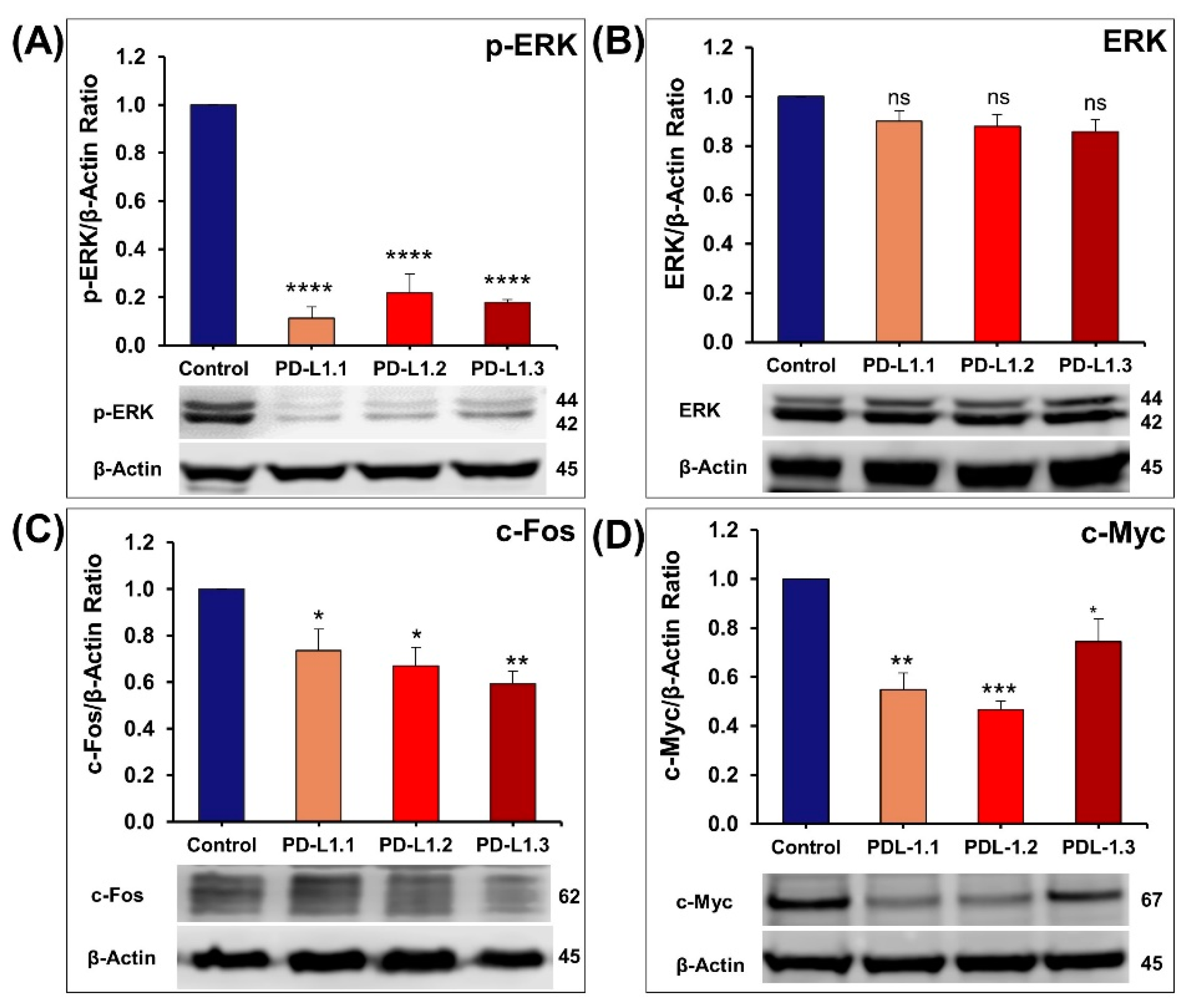
2.4.1. Impact of PD-L1 on ERK Signaling Pathway
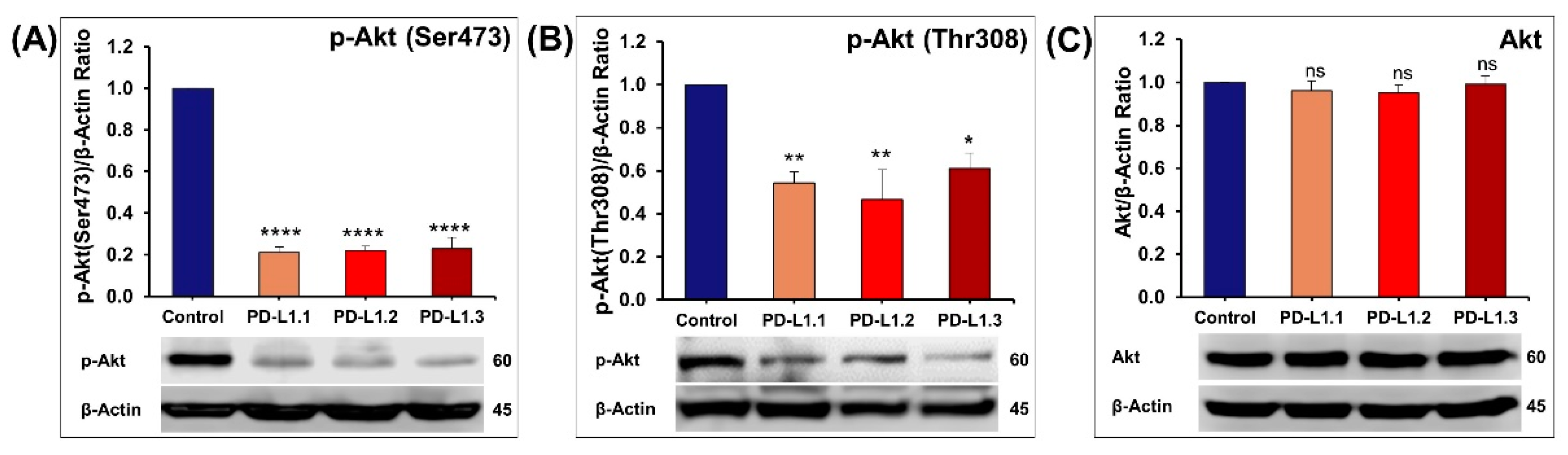
2.4.2. Impact of PD-L1 on Akt Signaling Pathway
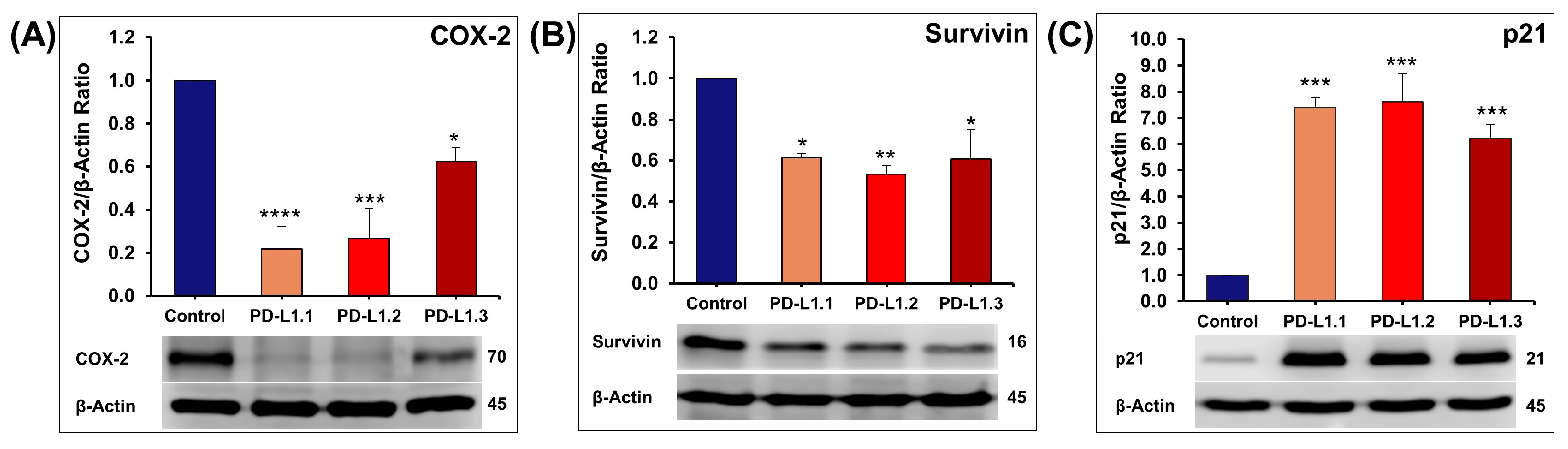
2.4.3. Investigation of ERK and Akt Downstream Proteins
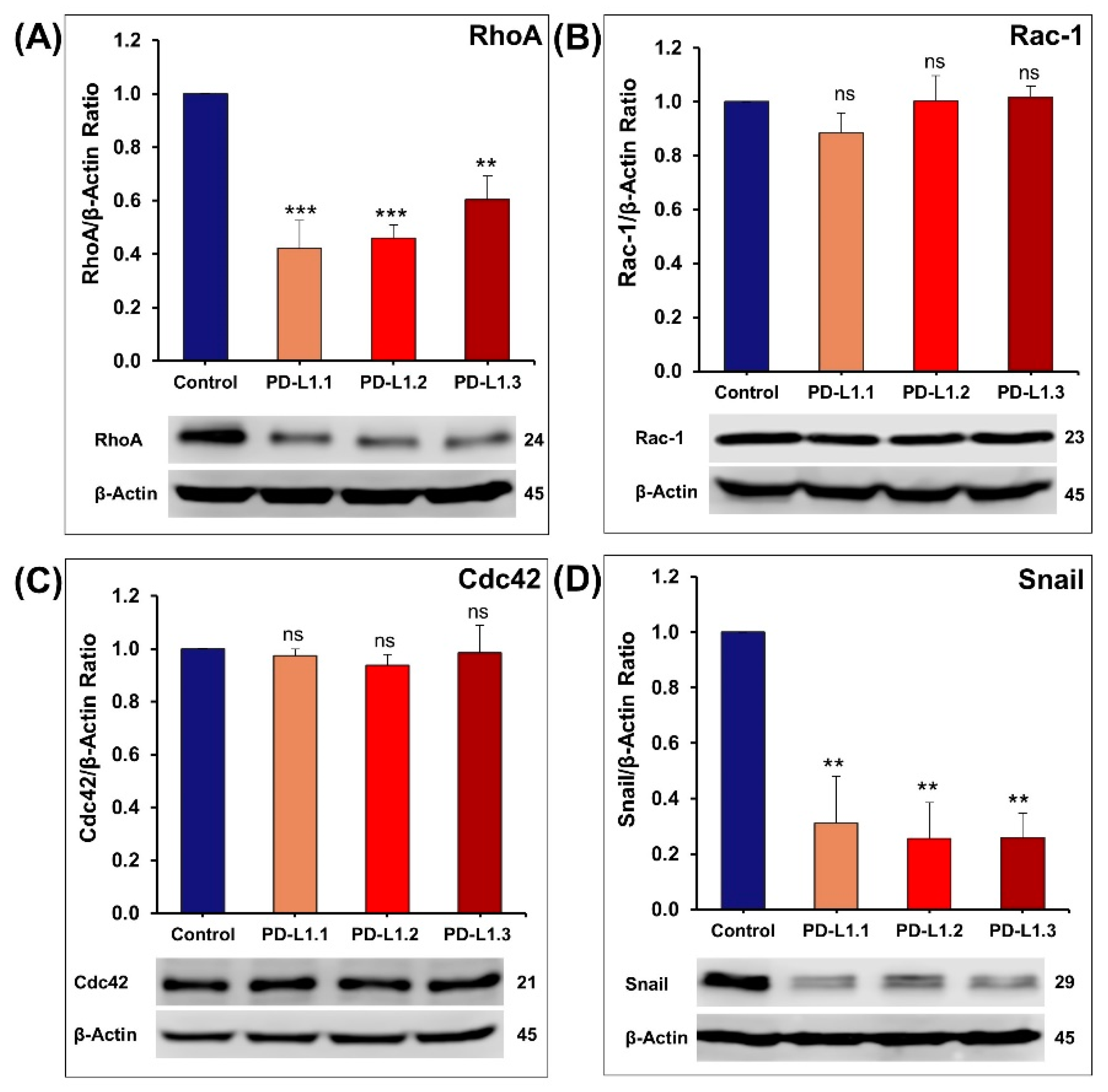
2.4.4. Impact of PD-L1 on Rho GTPases Signaling Pathway
3. Materials and Methods
3.1. Cell Culture and Antibodies
3.2. Establishment of Stable PD-L1 Knockout Clones in Breast Cancer Cells
3.3. Impact of PD-L1 Knockout on Cell Proliferation
3.4. Colony Formation Assay
3.5. Impact on Cellular Migration Using Wound Healing Assay
3.6. Boyden Chamber Matrigel Invasion Assay
3.7. The Oris™ Matrigel Cell Invasion Assay
3.8. Chick Embryo CAM Tumor Growth Assay
3.9. Western-Blot Analysis
3.10. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Ensenyat-Mendez, M.; Llinàs-Arias, P.; Orozco, J.I.J.; Íñiguez-Muñoz, S.; Salomon, M.P.; Sesé, B.; DiNome, M.L.; Marzese, D.M. Current Triple-Negative Breast Cancer Subtypes: Dissecting the Most Aggressive Form of Breast Cancer. Front. Oncol. 2021, 11, 681476. [Google Scholar] [CrossRef] [PubMed]
- Cirqueira, M.B.; Mendonça, C.R.; Noll, M.; Soares, L.R.; de Paula Carneiro Cysneiros, M.A.; Paulinelli, R.R.; Moreira, M.A.R.; Freitas-Junior, R. Prognostic Role of PD-L1 Expression in Invasive Breast Cancer: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 6090. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy—Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., III; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Xia, Y.; Wang, F.; Zhang, H.; Su, D.; Su, C.; Yang, C.; Wu, S.; An, S.; Lin, S.; et al. PD0325901, an ERK inhibitor, enhances the efficacy of PD-1 inhibitor in non-small cell lung carcinoma. Acta Pharm. Sin. B 2021, 11, 3120–3133. [Google Scholar] [CrossRef]
- Rom-Jurek, E.-M.; Kirchhammer, N.; Ugocsai, P.; Ortmann, O.; Wege, A.K.; Brockhoff, G. Regulation of Programmed Death Ligand 1 (PD-L1) Expression in Breast Cancer Cell Lines In Vitro and in Immunodeficient and Humanized Tumor Mice. Int. J. Mol. Sci. 2018, 19, 563. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Fang, Y.; Li, J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol. Lett. 2019, 18, 5399–5407. [Google Scholar] [CrossRef] [Green Version]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The Intracellular Signalosome of PD-L1 in Cancer Cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makuku, R.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. Current and Future Perspectives of PD-1/PDL-1 Blockade in Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 6661406. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727. [Google Scholar] [PubMed]
- Matikas, A.; Zerdes, I.; Lövrot, J.; Richard, F.; Sotiriou, C.; Bergh, J.; Valachis, A.; Foukakis, T. Prognostic Implications of PD-L1 Expression in Breast Cancer: Systematic Review and Meta-analysis of Immunohistochemistry and Pooled Analysis of Transcriptomic Data. Clin. Cancer Res. 2019, 25, 5717–5726. [Google Scholar] [CrossRef]
- Basu, G.D.; Ghazalpour, A.; Gatalica, Z.; Anderson, K.S.; McCullough, A.E.; Spetzer, D.B.; Pockaj, B.A. Expression of novel immunotherapeutic targets in triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 1001. [Google Scholar] [CrossRef]
- Li, M.; Li, A.; Zhou, S.; Xu, Y.; Xiao, Y.; Bi, R.; Yang, W. Heterogeneity of PD-L1 expression in primary tumors and paired lymph node metastases of triple negative breast cancer. BMC Cancer 2018, 18, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Shen, Y.; Zhu, X.; Lv, R.; Li, S.; Zhang, Z.; Shi, Y.G.; Tan, L. ERα is a negative regulator of PD-L1 gene transcription in breast cancer. Biochem. Biophys. Res. Commun. 2018, 505, 157–161. [Google Scholar] [CrossRef]
- Ghebeh, H.; Tulbah, A.; Mohammed, S.; Elkum, N.; Bin Amer, S.M.; Al-Tweigeri, T.; Dermime, S. Expression of B7-H1 in breast cancer patients is strongly associated with high proliferative Ki-67-expressing tumor cells. Int. J. Cancer 2007, 121, 751–758. [Google Scholar] [CrossRef]
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 Tumor-Intrinsic Signaling and Its Therapeutic Implication in Triple-Negative Breast Cancer. JCI Insight. 2021, 6, e131458. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Xiong, Y.; Zheng, X.; Xie, Q.; Zhou, Q.; Shi, L.; Wu, C.; Jiang, J.; Wang, H. Knockdown of PD-L1 in Human Gastric Cancer Cells Inhibits Tumor Progression and Improves the Cytotoxic Sensitivity to CIK Therapy. Cell. Physiol. Biochem. 2017, 41, 907–920. [Google Scholar] [CrossRef]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xiong, Y.; Li, J.; Zheng, X.; Zhou, Q.; Turner, A.; Wu, C.; Lu, B.; Jiang, J. PD-L1 Expression Promotes Epithelial to Mesenchymal Transition in Human Esophageal Cancer. Cell. Physiol. Biochem. 2017, 42, 2267–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geum, D.-H.; Hwang, D.-S.; Lee, C.-H.; Cho, S.-D.; Jang, M.-A.; Ryu, M.-H.; Kim, U.-K. PD-L1 Expression Correlated with Clinicopathological Factors and Akt/Stat3 Pathway in Oral SCC. Life 2022, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Lotfinejad, P.; Kazemi, T.; Safaei, S.; Amini, M.; Roshani Asl, E.; Baghbani, E.; Sandoghchian Shotorbani, S.; Jadidi Niaragh, F.; Derakhshani, A.; Abdoli Shadbad, M.; et al. PD-L1 silencing inhibits triple-negative breast cancer development and upregulates T-cell-induced pro-inflammatory cytokines. Biomed. Pharmacother. 2021, 138, 111436. [Google Scholar] [CrossRef]
- Yu, W.; Hua, Y.; Qiu, H.; Hao, J.; Zou, K.; Li, Z.; Hu, S.; Guo, P.; Chen, M.; Sui, S.; et al. PD-L1 promotes tumor growth and progression by activating WIP and β-catenin signaling pathways and predicts poor prognosis in lung cancer. Cell Death Dis. 2020, 11, 506. [Google Scholar] [CrossRef]
- Wang, Y.; Rousset, X.; Prunier, C.; Garcia, P.; Dosda, E.; Leplus, E.; Viallet, J. PD-1/PD-L1 Checkpoint Inhibitors Are Active in the Chicken Embryo Model and Show Antitumor Efficacy In Ovo. Cancers 2022, 14, 3095. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Hu, D.X.; Chen, W.-Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Eichberger, J.; Schulz, D.; Pscheidl, K.; Fiedler, M.; Reichert, T.; Bauer, R.; Ettl, T. PD-L1 Influences Cell Spreading, Migration and Invasion in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8089. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, L.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; et al. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget 2017, 8, 30276–30287. [Google Scholar] [CrossRef] [Green Version]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Passariello, M.; D’Alise, A.M.; Esposito, A.; Vetrei, C.; Froechlich, G.; Scarselli, E.; Nicosia, A.; De Lorenzo, C. Novel Human Anti-PD-L1 mAbs Inhibit Immune-Independent Tumor Cell Growth and PD-L1 Associated Intracellular Signalling. Sci. Rep. 2019, 9, 13125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, R.; Taha, R.Z.; Sasidharan Nair, V.; Alajez, N.M.; Elkord, E. PD-L1 Blockade by Atezolizumab Downregulates Signaling Pathways Associated with Tumor Growth, Metastasis, and Hypoxia in Human Triple Negative Breast Cancer. Cancers 2019, 11, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazon, H.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M., Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front. Microbiol. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, P.; Hernández-Losa, J.; Lyons, R.J.; Castellone, M.D.; Gutkind, J.S. Regulation of the Transcriptional Activity of c-Fos by ERK. J. Biol. Chem. 2005, 280, 35081–35084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Ren, X.; Du, Y.; Zhao, W.; Cui, M.; Chen, G.; Zhao, Y. c-Fos/ERK promotes the progression from pancreatic intraepithelial neoplasia to pancreatic ductal adenocarcinoma. Oncol. Rep. 2016, 36, 3413–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Gupta, N.; Jung, K.; Wu, C.; Alshareef, A.; Alqahtani, H.; Damaraju, S.; Mackey, J.R.; Ghosh, S.; Sabri, S.; Abdulkarim, B.S.; et al. High Myc expression and transcription activity underlies intra-tumoral heterogeneity in triple-negative breast cancer. Oncotarget 2017, 8, 28101–28115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Qiao, M.; Iglehart, J.D.; Pardee, A.B. Metastatic Potential of 21T Human Breast Cancer Cells Depends on Akt/Protein Kinase B Activation. Cancer Res 2007, 67, 5293–5299. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Zhang, T.; Deng, S.-C.; Wei, J.-C.; Yang, P.; Wang, Q.; Chen, Z.-P.; Li, W.-L.; Chen, H.-C.; Hu, H.; et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/AkT/MTOR signaling pathway. OncoTargets Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Z.; Deng, Z.; Zhou, L.; Li, K.; Xia, X.; Xie, R. PD-L1 Induces Epithelial-Mesenchymal Transition in Nasopharyngeal Carcinoma Cells Through Activation of the PI3K/AKT Pathway. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2019, 27, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef] [Green Version]
- A Glynn, S.; Prueitt, R.L.; A Ridnour, L.; Boersma, B.J.; Dorsey, T.M.; A Wink, D.; E Goodman, J.; Yfantis, H.G.; Lee, D.H.; Ambs, S. COX-2 activation is associated with Akt phosphorylation and poor survival in ER-negative, HER2-positive breast cancer. BMC Cancer 2010, 10, 626. [Google Scholar] [CrossRef] [Green Version]
- Chi, F.; Wu, R.; Jin, X.; Jiang, M.; Zhu, X. HER2 induces cell proliferation and invasion of non-small-cell lung cancer by upregulating COX-2 expression via MEK/ERK signaling pathway. OncoTargets Ther. 2016, 9, 2709–2716. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-S.; Hellmich, M.R.; Wen, X.D.; Townsend, C.M. Activator Protein-1 Transcription Factor Mediates Bombesin-stimulated Cyclooxygenase-2 Expression in Intestinal Epithelial Cells. J. Biol. Chem. 2001, 276, 22941–22947. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, J.A.; Liu, T.; Goodson, A.G.; Grossman, D. Survivin Enhances Motility of Melanoma Cells by Supporting Akt Activation and α5 Integrin Upregulation. Cancer Res. 2010, 70, 7927–7937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Meng, Q.; Liu, L.-Z.; You, Y.-P.; Liu, N.; Jiang, B.-H. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem. Biophys. Res. Commun. 2010, 395, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-P.; Dong, X.; Lin, L.; Jiang, X.; Wei, Z.; Zhai, B.; Sun, B.; Zhang, Q.; Wang, X.; Jiang, H.; et al. Up-regulation of survivin by AKT and hypoxia-inducible factor 1α contributes to cisplatin resistance in gastric cancer. FEBS J. 2013, 281, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Haywood, P.; Flint, P.; Knox, W.F.; Bundred, N.J. Survivin expression in in situ and invasive breast cancer relates to COX-2 expression and DCIS recurrence. Br. J. Cancer 2006, 94, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysan, K.; Merchant, F.H.; Zhu, L.; Dohadwala, M.; Luo, J.; Lin, Y.; Heuze-Vourc'H, N.; Põld, M.; Seligson, D.; Chia, D.; et al. COX-2-dependent stabilization of survivin in non-small cell lung cancer. FASEB J. 2003, 18, 206–208. [Google Scholar] [CrossRef]
- Sato, A.; Mizobuchi, Y.; Nakajima, K.; Shono, K.; Fujihara, T.; Kageji, T.; Kitazato, K.; Matsuzaki, K.; Mure, H.; Kuwayama, K.; et al. Blocking COX-2 induces apoptosis and inhibits cell proliferation via the Akt/survivin- and Akt/ID3 pathway in low-grade-glioma. J. Neuro-Oncol. 2017, 132, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hao, Q.; Cao, W.; Vadgama, J.V.; Wu, Y. Celecoxib in breast cancer prevention and therapy. Cancer Manag. Res. 2018, 10, 4653–4667. [Google Scholar] [CrossRef] [Green Version]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor P21cip1/Waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [Green Version]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Suh, E.-J.; Kim, Y.-J.; Kim, S.H. Protein phosphatase 2Cγ regulates the level of p21Cip1/WAF1 by Akt signaling. Biochem. Biophys. Res. Commun. 2009, 386, 467–470. [Google Scholar] [CrossRef]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.-H.; Hung, M.-C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.-D.; Zhao, Y.-J.; Jia, X.-Y.; Wu, J.; Wang, Y.-G.; Huang, F. Multifaceted p21 in carcinogenesis, stemness of tumor and tumor therapy. World J. Stem Cells 2020, 12, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.; Herraizy, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e983867. [Google Scholar] [CrossRef] [PubMed]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in Cancer: Friend or Foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. Rho GTPases: Big Players in Breast Cancer Initiation, Metastasis and Therapeutic Responses. Cells 2020, 9, 2167. [Google Scholar] [CrossRef]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C.; et al. Anti-RhoA and Anti-RhoC siRNAs Inhibit the Proliferation and Invasiveness of MDA-MB-231 Breast Cancer Cells in Vitro and in Vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef]
- Liberto, M.; Cobrinik, D.; Minden, A. Rho regulates p21CIP1, cyclin D1, and checkpoint control in mammary epithelial cells. Oncogene 2002, 21, 1590–1599. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Wang, Q.; Han, Z.; Hu, W.; Xi, L.; Gao, Q.; Wang, S.; Zhou, J.; Xu, G.; Meng, L.; et al. Reduced expression of Snail decreases breast cancer cell motility by downregulating the expression and inhibiting the activity of RhoA GTPase. Oncol. Lett. 2013, 6, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Koh, J.; Kim, M.-Y.; Kwon, D.; Go, H.; A Kim, Y.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef]
- Al-Azawi, A.; Sulaiman, S.; Arafat, K.; Yasin, J.; Nemmar, A.; Attoub, S. Impact of Sodium Dichloroacetate Alone and in Combination Therapies on Lung Tumor Growth and Metastasis. Int. J. Mol. Sci. 2021, 22, 12553. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkaabi, D.; Arafat, K.; Sulaiman, S.; Al-Azawi, A.M.; Attoub, S. PD-1 Independent Role of PD-L1 in Triple-Negative Breast Cancer Progression. Int. J. Mol. Sci. 2023, 24, 6420. https://doi.org/10.3390/ijms24076420
Alkaabi D, Arafat K, Sulaiman S, Al-Azawi AM, Attoub S. PD-1 Independent Role of PD-L1 in Triple-Negative Breast Cancer Progression. International Journal of Molecular Sciences. 2023; 24(7):6420. https://doi.org/10.3390/ijms24076420
Chicago/Turabian StyleAlkaabi, Duaa, Kholoud Arafat, Shahrazad Sulaiman, Aya Mudhafar Al-Azawi, and Samir Attoub. 2023. "PD-1 Independent Role of PD-L1 in Triple-Negative Breast Cancer Progression" International Journal of Molecular Sciences 24, no. 7: 6420. https://doi.org/10.3390/ijms24076420