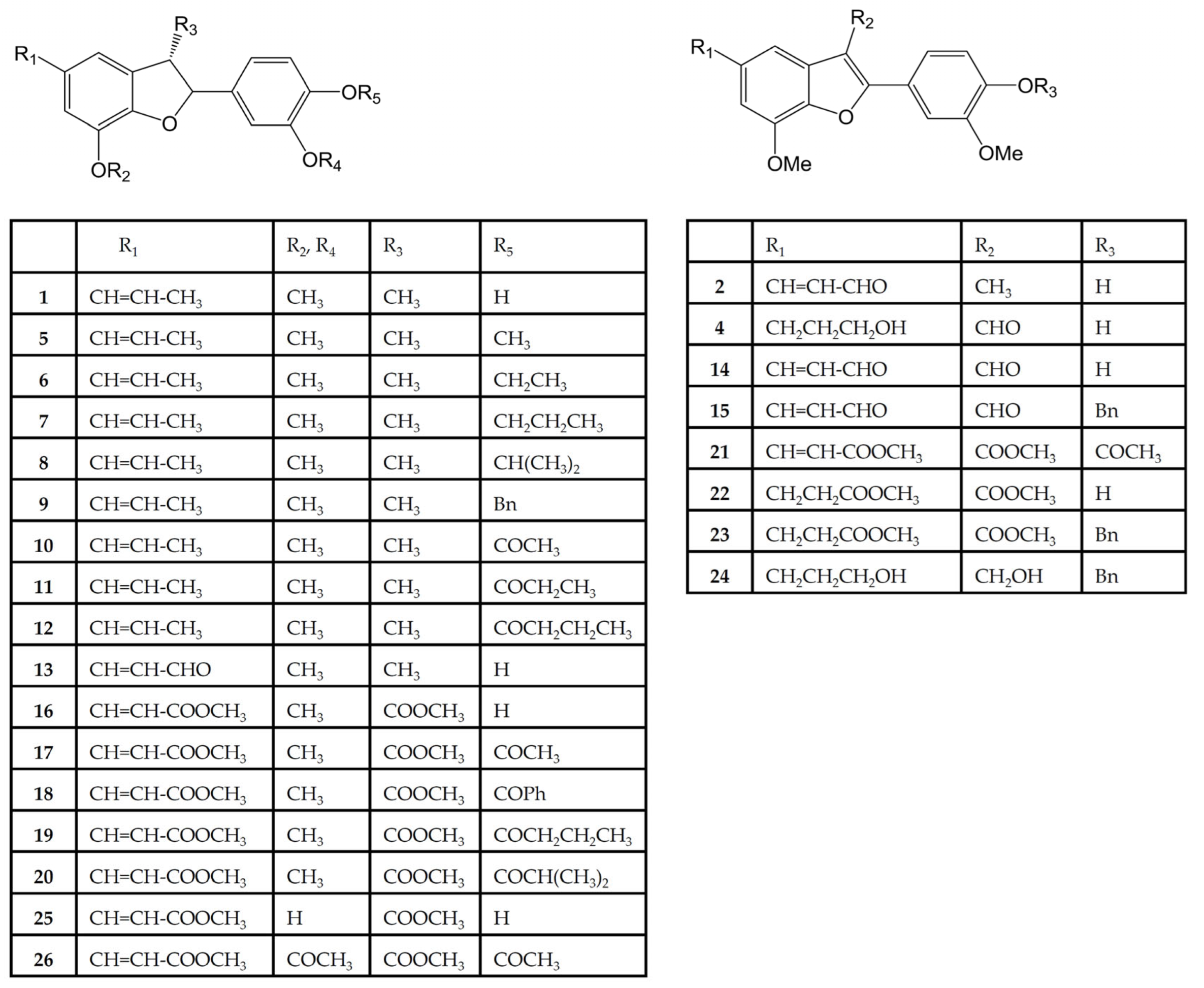
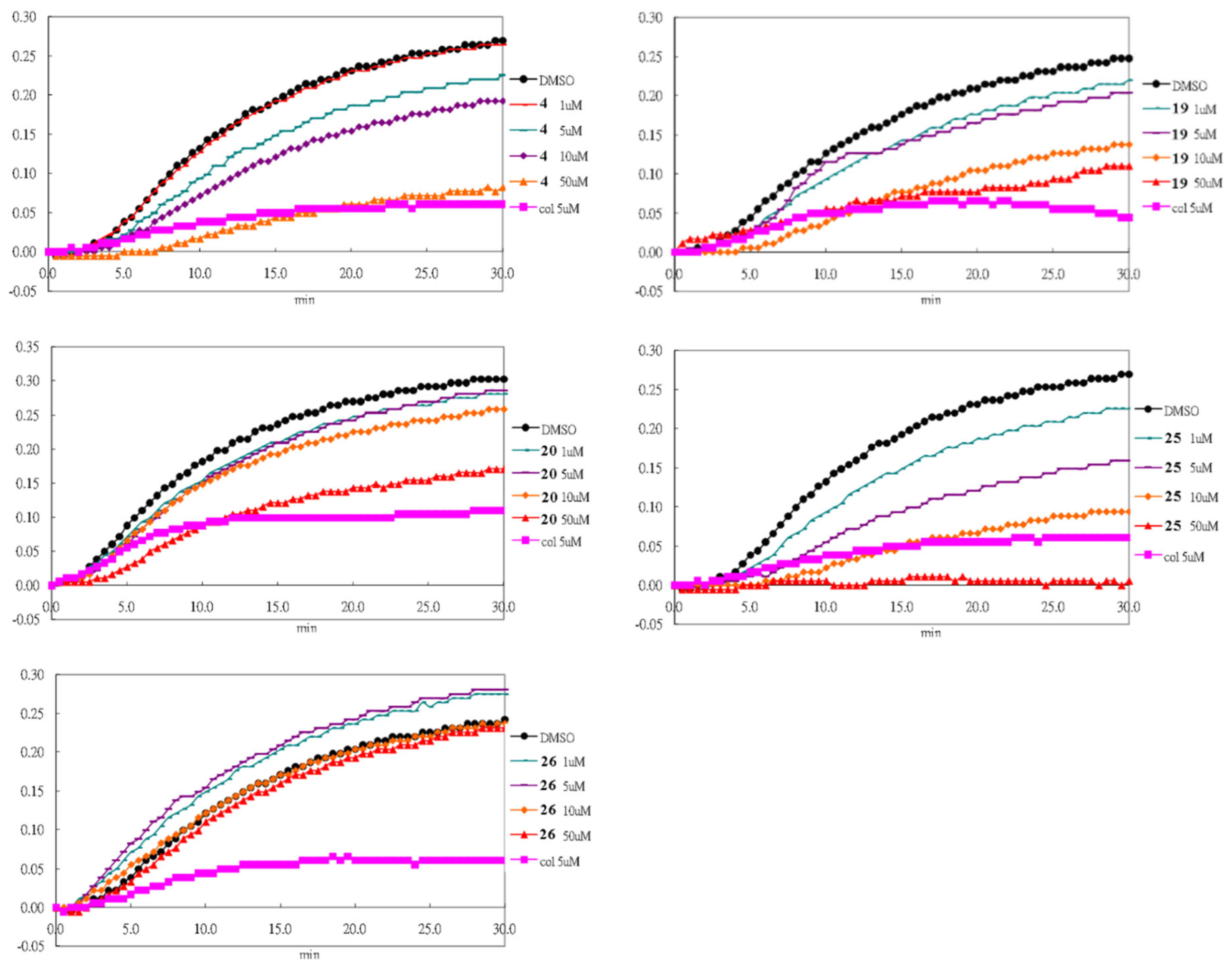
3.2. Chemistry
The salvinal derivatives were synthesized at the laboratory of Professor Yueh-Hsiung Kuo of the Tsuzuki Institute for Traditional Medicine, China Medical University (Taichung, Taiwan).
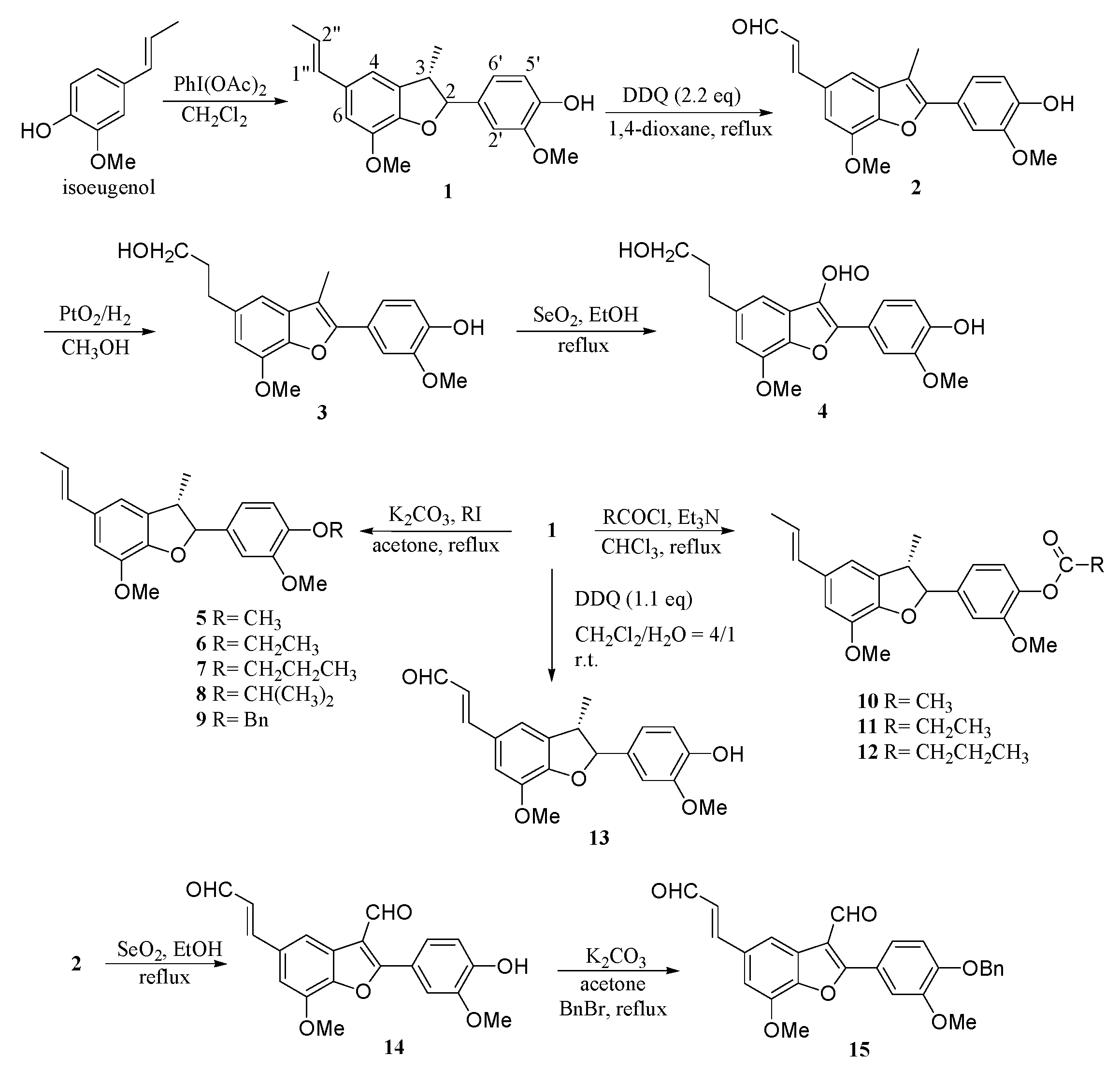
Compound 1 was prepared from isoeugenol by using IDA (iodobenzene diacetate). The solution of isoeugenol (10.0 g in 100 mL of CH2Cl2) was added dropwise to the solution of IDA (10.0 g, 30.2 mmol) in 100 mL of CH2Cl2 (dry with CaH2) at room temperature for 4 h. After 48 h, NaHCO3 (3 g) was added to the solution and stirred for 1 h. The mixture was filtrated, and the filtrate was evaporated under reduced pressure to give a yellow oil. Then, the residue was purified by Si gel column chromatography to give 1 as a colorless solid (3.9 g, 40% yield; with solvent system EtOAc: hexane = 1:9); mp 123–124 °C; 1H-NMR (CDCl3) δH 6.96 (s, 1H, H-4), 6.88 (d, J = 8.1 Hz, 1H, H-6′), 6.86 (d, J = 8.1 Hz, 1H, H-5′), 6.77 (s, 1H, H-6), 6.75 (s, 1H, H-2′), 6.35 (dd, J = 15.6, 1.0 Hz, 1H, H-1″), 6.09 (dq, J = 15.6, 6.6 Hz, 1H, H-2″), 5.64 (s, 1H, 4′-OH), 5.08 (d, J = 9.5 Hz, 1H, H-2), 3.87 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.43 (m, 1H, H-3), 1.85 (dd, J = 6.6, 1.0 Hz, 3H, H-3″), 1.36 (d, J = 6.8 Hz, 3H, Me-C-3); 13C-NMR (CDCl3) δC 146.5 (C-7), 146.4 (C-3′), 145.6 (C-7a), 143.9 (C-4′), 137.2 (C-1′), 132.0 (C-3a), 131.9 (C-5), 130.8 (C-1″), 123.2 (C-6′), 119.7 (C-2″), 114.0 (C-4), 113.2 (C-2′), 109.1 (C-5′), 108.8 (C-6), 93.6 (C-2), 55.7 (C-7-OMe), 51.7 (C-3′-OMe), 45.4 (C-3), 18.2 (C-3″), 17.4 (C-3-Me); IR (KBr film) νmax 3446, 3023, 2964, 1608, 1515, 1460, 1337, 1275, 1141, 1033, 962 cm−1; EI-MS m/z (%) (70 eV) 326 (M+, 45; C20H22O4), 202 (30), 178 (45), 151 (100), 137 (8), 119 (11), 91 (15).
Compound 2 was prepared from 1 by using DDQ (dichlorodicyanobenzoquinone). Compound 1 (2.12 g) and DDQ (3.24 g) were resolved in 50 mL of 1,4-dioxane. The solution was then refluxed. After 48 h, the solution was filtrated, and the filtrate was evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give 2 as a colorless solid (1.81 g, 83% yield; with solvent system EtOAc: hexane = 1:8); mp 221–222 °C; 1H-NMR (CDCl3) δH 9.68 (d, J = 7.6 Hz, 1H, H-3″), 7.55 (d, J = 15.9 Hz, 1H, H-1″), 7.26–7.30 (m, 3H, H-4, H-6, H-6′), 6.99 (d, J = 8.0 Hz, 1H, H-5′), 6.97 (s, 1H, H-2′), 6.70 (dd, J = 15.9, 7.6 Hz, 1H, H-2″), 5.85 (br s, 1H, Ph-OH), 4.03 (s, 3H, MeO-C-7), 3.95 (s, 3H, MeO-C-3′), 2.40 (s, 3H, Me-C-3); 13C-NMR (CDCl3) δC 193.6, 153.9, 146.7, 146.2, 145.2, 144.6, 133.5, 129.6, 127.4, 123.0, 120.8, 114.6, 113.8, 110.1, 109.4, 105.6, 56.1, 9.5; IR (KBr film) νmax 3540, 2949, 2811, 2727, 1674, 1616, 1513, 1214, 1128, 972 cm−1; EI-MS m/z (%) (70 eV) 338 (M+, 100; C20H18O5), 326 (21), 310 (100), 295 (14), 267 (15), 151 (13), 137 (14), 69 (17), 57 (19).
Compound 3 was prepared from 2 using Adam’s catalyst reduction reaction. The solution of 2 (1.33 g in 20 mL of CH3OH) with 10% PtO2/H2O (96.3 mg) was stirred under H2 at room temperature. After 6 h, the mixture was filtrated, and the filtrate was evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give 3 as a colorless solid (1.28 g, 95% yield; with solvent system EtOAc: hexane = 2:5); mp 165–166 °C; 1H-NMR (CDCl3) δH 7.30 (d, J = 2.0 Hz, 1H, H-4), 7.26 (dd, J = 8.2, 1.6 Hz, 1H, H-6′), 6.98 (d, J = 8.2 Hz, 1H, H-5′), 6.91 (d, J = 1.0 Hz, 1H, H-2′), 6.63 (d, J = 2.0 Hz, 1H, H-6), 5.73 (s, 1H, Ph-OH), 4.01 (s, 3H, MeO-C-7), 3.96 (s, 3H, MeO-C-3′), 3.71 (t, J = 6.5 Hz, 2H, H-3″), 2.79 (t, J = 7.9 Hz, 2H, H-1″), 2.38 (s, 3H, Me-C-3), 1.95 (m, 2H, H-2″); 13C-NMR (CDCl3) δC 151.4, 146.6, 145.7, 144.7, 136.9, 133.0, 123.8, 120.6, 114.5, 110.8, 109.5, 107.4, 62.4, 56.1, 34.8, 32.6, 9.60; IR (KBr film) νmax 3431, 2940, 2851, 1602, 1516, 1455, 1386, 1222, 1052, 793 cm−1; EI-MS m/z (%) (70 eV) 342 (M+, 5; C20H22O5), 340 (100), 324 (37), 312 (26), 297 (19), 284 (20), 148 (13), 97 (16), 91 (18), 69 (23), 57 (28).
Compound
4 was prepared from
3 using SeO
2 oxidative reaction. The solution of
3 (0.64 g in 20 mL of EtOH) with SeO
2 (0.42 g) was refluxed. After 12 h, the mixture was evaporated under reduced pressure. Then, 30 mL of EtOAc was added, the mixture was filtrated by celite, and the filtrate was evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give
4 as a colorless solid (0.48 g, 72% yield; with solvent system EtOAc: hexane = 1:5); mp 173–174 °C;
1H-NMR (CDCl
3) δ
H 10.25 (s, 1H, CHO), 7.64 (s, 1H, H-2′), 7.37 (d, J = 8.0 Hz, 1H, H-6′), 7.35 (s, 1H, H-4), 7.04 (d, J = 8.0 Hz, 1H, H-5′), 6.73 (s, 1H, H-6), 6.11 (br s, 1H, Ph-OH), 4.00 (s, 3H, OMe), 3.97 (s, 3H, OMe), 3.69 (t, J = 6.5 Hz, 2H, H-3″), 2.80 (t, J = 7.3 Hz, 2H, H-1″), 1.94 (m, 2H, H-2″);
13C-NMR (CDCl
3) δ
C 186.8, 165.9, 148.7, 146.9, 144.6, 141.6, 139.9, 127.3, 123.7, 120.6, 116.7, 115.0, 113.5, 111.0, 108.8, 62.2, 56.3, 56.1, 34.7, 32.5; IR (KBr film) ν
max 3513, 3435, 2940, 2864, 1637, 1601, 1522, 1490, 1409, 1273, 1139, 1061, 818 cm
−1; EI-MS
m/
z (%) (70 eV) 356 (M
+, 60; C
20H
20O
6), 312 (100), 269 (7), 197 (6), 152 (6), 137 (6), 126 (4), 105 (4), 91 (4), 55 (4) (
Figures S1–S4).
Compound 5 was prepared from 1 using alkylation reaction. MeI (66.2 mg) and K2CO3 (100.3 mg) were added to the solution of 1 (100.4 mg, in 10 mL of acetone), and then the solution was refluxed. After 6 h, the mixture was filtrated, and the filtrate was evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give 5 as a colorless solid (92.8 mg, 88% yield; with solvent system EtOAc: hexane = 1:9); mp 118–119 °C; 1H-NMR (CDCl3) δH 6.96 (s, 1H, H-4), 6.94 (dd, J = 8.1, 1.7 Hz, 1H, H-6′), 6.82 (d, J = 8.1 Hz, 1H, H-5′), 6.77 (s, 1H, H-6), 6.75 (s, 1H, H-2′), 6.34 (br d, J = 15.8 Hz, 1H, H-1″), 6.09 (dq, J = 15.8, 6.6 Hz, 1H, H-2″), 5.09 (d, J = 9.5 Hz, 1H, H-2), 3.87 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.85 (s, 3H, OMe), 3.44 (m, 1H, H-3), 1.85 (dd, J = 6.6, 1.3 Hz, 3H, H-3″), 1.36 (d, J = 6.9 Hz, 3H, Me-C-3); 13C-NMR (CDCl3) δC 149.1, 146.5, 144.1, 137.4, 133.2, 132.6, 132.2, 130.9, 130.2, 127.4, 123.4, 119.2, 113.3, 110.8, 109.5, 109.2, 93.6, 55.9, 45.5, 18.3, 17.6; IR (KBr film) νmax 3014, 2963, 2882, 1604, 1517, 1464, 1270, 1151, 1023, 957, 855, 817 cm−1; EI-MS m/z (%) (70 eV) 340 (M+, 28; C21H24O4), 204 (25), 192 (40), 168 (28), 165 (100), 153 (27), 125 (13), 81 (14), 77 (23), 69 (29).
Compound 6 was prepared from 1 using alkylation reaction; the reaction was similar to the preparation of 5. Compound 1 (100.4 mg) gave 6 as a colorless solid (93.4 mg, 85% yield; with solvent system EtOAc: hexane = 1:9); mp 106–107 °C; 1H-NMR (CDCl3) δH 6.96 (s, 1H, H-4), 6.91 (dd, J = 8.1, 1.9 Hz, 1H, H-6′), 6.82 (d, J = 8.1 Hz, 1H, H-5′), 6.77 (s, 1H, H-6), 6.75 (d, J = 1.9 Hz, 1H, H-2′), 6.35 (dq, J = 15.6, 1.4 Hz, 1H, H-1″), 6.09 (dq, J = 15.6, 6.2 Hz, 1H, H-2″), 5.09 (d, J = 9.3 Hz, 1H, H-2), 4.08 (q, J = 6.9 Hz, 2H, OCH2CH3), 3.87 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.45 (m, 1H, H-3), 1.85 (dd, J = 6.2, 1.4 Hz, 3H, H-3″), 1.44 (t, J = 6.9 Hz, 3H, OCH2CH3), 1.37 (d, J = 6.7 Hz, 3H, Me-C-3); 13C-NMR (CDCl3) δC 149.4, 148.4, 146.5, 144.1, 137.4, 133.2, 132.5, 132.1, 130.9, 130.2, 127.4, 123.4, 119.2, 113.3, 112.3, 109.8, 109.2, 93.6, 64.3, 55.9, 55.8, 45.5, 18.3, 17.6, 14.7; IR (KBr film) νmax 3011, 2965, 2884, 1600, 1517, 1461, 1339, 1227, 1031, 957, 856 cm−1; EI-MS m/z (%) (70 eV) 354 (M+, 14; C22H26O4), 266 (14), 206 (80), 179 (100), 151 (89), 119 (10), 91 (17), 77 (13).
Compound 7 was prepared from 1 using alkylation reaction; the reaction was similar to the preparation of 5. Compound 1 (100.4 mg) afforded 7 as a colorless solid (94.8 mg, 83% yield; with solvent system EtOAc: hexane = 1:9); mp 109–110 °C; 1H-NMR (CDCl3) δH 6.96 (s, 1H, H-4), 6.91 (dd, J = 8.1, 1.1 Hz, 1H, H-6′), 6.81 (d, J = 8.1 Hz, 1H, H-5′), 6.76 (s, 1H, H-6), 6.75 (s, 1H, H-2′), 6.35 (dq, J = 15.6, 1.0 Hz, 1H, H-1″), 6.09 (dq, J = 15.6, 6.6 Hz, 1H, H-2″), 5.09 (d, J = 9.5 Hz, 1H, H-2), 3.95 (t, J = 6.4 Hz, 2H, OCH2CH2CH3), 3.87 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.44 (m, 1H, H-3), 1.78–1.88 (m, 5H, H-3″, OCH2CH2CH3), 1.33 (d, J = 6.4 Hz, 3H, Me-C-3), 1.01 (t, J = 7.4 Hz, 3H, OCH2CH2CH3); 13C-NMR (CDCl3) δC 149.4, 148.6, 146.5, 144.1, 133.2, 132.5, 132.1, 130.9, 130.2, 123.3, 119.1, 113.2, 112.5, 109.9, 109.2, 93.6, 70.4, 55.9, 55.8, 45.5, 29.6, 22.4, 18.3, 17.5, 10.3; IR (KBr film) νmax 2965, 2884, 1600, 1518, 1462, 1267, 1145, 1031, 957, 856, 807 cm−1; EI-MS m/z (%) (70 eV) 368 (M+, 27; C23H28O4), 280 (12), 220 (40), 193 (44), 178 (23), 151 (100), 140 (33), 97 (32), 77 (44), 57 (63).
Compound 8 was prepared from 1 using alkylation reaction; the reaction was similar to the preparation of 5. Compound 1 (100.4 mg) obtained 8 as a colorless solid purified on SiO2 column chromatography (114.2 mg, 82% yield; with solvent system EtOAc: hexane = 1:9); mp 107–108 °C; 1H-NMR (CDCl3) δH 6.96 (s, 1H, H-4), 6.91 (dd, J = 8.1, 1.1 Hz, 1H, H-6′), 6.84 (d, J = 8.1 Hz, 1H, H-5′), 6.77 (s, 1H, H-6), 6.75 (d, J = 1.1 Hz, 1H, H-2′), 6.35 (d, J = 15.6 Hz, 1H, H-1″), 6.09 (dq, J = 15.6, 6.5 Hz, 1H, H-2″), 5.09 (d, J = 9.4 Hz, 1H, H-2), 4.50 (m, 1H, OCH(CH3)2), 3.87 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.46 (m, 1H, H-3), 1.85 (d, J = 6.5 Hz, 3H, H-3″), 1.36 (d, J = 7.7 Hz, 3H, Me-C-3), 1.34 (d, J = 6.3 Hz, 6H, OCH(CH3)2); 13C-NMR (CDCl3) δC 150.4, 147.3, 144.9, 133.2, 132.9, 132.1, 130.9, 123.3, 119.1, 115.4, 113.2, 110.3, 109.2, 93.6, 71.4, 56.0, 55.9, 55.8, 45.4, 21.9, 18.3, 17.6; IR (KBr film) νmax 2975, 2935, 1603, 1508, 1461, 1333, 1269, 1138, 1035, 958, 856, 820 cm−1; EI-MS m/z (%) (70 eV) 368 (M+, 5; C23H28O4), 326 (8), 280 (10), 220 (12), 178 (59), 151 (100), 140 (28), 91 (9), 71 (8), 57 (13).
Compound 9 was prepared from 1 using alkylation reaction; the reaction was similar to the preparation of 5. Compound 1 (100.4 mg) yielded 9 as a colorless solid on SiO2 column chromatography (103.3 mg, 80% yield; with solvent system EtOAc: hexane = 1:9); mp 165–166 °C; 1H-NMR (CDCl3) δH 7.27–7.42 (m, 5H, OCH2Ph), 6.98 (s, 1H, H-4), 6.86 (dd, J = 8.2, 1.5 Hz, 1H, H-6′), 6.82 (d, J = 8.2 Hz, 1H, H-5′), 6.76 (s, 1H, H-6), 6.74 (d, J = 1.5 Hz, 1H, H-2′), 6.34 (dq, J = 16.0, 1.2 Hz, 1H, H-1″), 6.05 (dq, J = 16.0, 6.7 Hz, 1H, H-2″), 5.14 (s, 2H, OCH2Ph), 5.08 (d, J = 9.6 Hz, 1H, H-2), 3.87 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.44 (m, 1H, H-3), 1.85 (dd, J = 6.7, 1.2 Hz, 3H, H-3″), 1.35 (d, J = 6.8 Hz, 3H, Me-C-3); 13C-NMR (CDCl3) δC 149.7, 148.1, 146.5, 144.0, 137.0, 133.1, 132.1, 130.8, 128.4, 127.7, 127.1, 123.3, 119.0, 113.6, 113.2, 110.0, 109.2, 93.5, 70.9, 55.9, 55.8, 45.4, 18.3, 17.6; IR (KBr film) νmax 3069, 3002, 2961, 2871, 1602, 1516, 1460, 1268, 1220, 1141, 1031, 947, 742 cm−1; EI-MS m/z (%) (70 eV) 416 (M+, 3; C27H28O4), 328 (8), 268 (25), 241 (10), 177 (42), 151 (32), 139 (14), 91 (100), 65 (8).
Compound 10 was prepared from 1 using esteration reaction. CH3COCl (0.3 mL) and Et3N (0.4 mL) were added to the solution of 1 (107.6 mg in 10mL of CHCl3), and then the solution was refluxed. After 3 h, the mixture was added to ice water (10 mL) and extracted by EtOAc (2 × 10 mL). The organic layers were combined and then washed with 1N HCl and aqueous NaHCO3 and subsequently evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give 10 as a colorless solid (117.4 mg, 92% yield; with solvent system EtOAc: hexane = 1:9); mp 154–155 °C; 1H-NMR (CDCl3) δH 7.04 (s, 1H, H-4), 7.02 (d, J = 8.2 Hz, 1H, H-5′), 6.94 (dd, J = 8.2, 1.7 Hz, 1H, H-6′), 6.77 (s, 1H, H-6), 6.74 (s, 1H, H-2′), 6.34 (dq, J = 15.4, 1.1 Hz, 1H, H-1″), 6.09 (dq, J = 15.4, 6.6 Hz, 1H, H-2″), 5.14 (d, J = 9.1 Hz, 1H, H-2), 3.88 (s, 3H, OMe), 3.80 (s, 3H, OMe), 3.46 (m, 1H, H-3), 2.29 (s, 3H, OAc), 1.85 (dd, J = 6.6, 1.1 Hz, 3H, H-3″), 1.38 (d, J = 6.7 Hz, 3H, Me-C-3); 13C-NMR (CDCl3) δC 168.9, 151.2, 146.4, 144.1, 139.6, 139.2, 133.0, 132.3, 130.8, 123.6, 122.6, 118.6, 113.3, 110.2, 109.3, 93.0, 55.9, 55.8, 45.7, 20.6, 18.3, 17.9; IR (KBr film) νmax 2968, 2938, 2881, 1769, 1608, 1507, 1461, 1202, 1152, 1035, 966, 860 cm−1; EI-MS m/z (%) (70 eV) 368 (M+, 37; C22H24O5), 326 (100), 182 (19), 172 (38), 140 (60), 127 (42), 98 (23), 85 (46), 71 (24), 57 (29).
Compound 11 was prepared from 1 using esteration reaction; the reaction was similar to the preparation of 10 with propanoyl chloride and triethylamine. Compound 1 (121.1 mg) afforded 11 as a colorless solid (133.6 mg, 95% yield; with solvent system EtOAc: hexane = 1:9); mp 175–176 °C; 1H-NMR (CDCl3) δH 7.03 (s, 1H, H-4), 6.98 (d, J = 8.0 Hz, 1H, H-5′), 6.94 (dd, J = 8.0, 1.4 Hz, 1H, H-6′), 6.77 (s, 1H, H-6), 6.74 (d, J = 1.4 Hz, 1H, H-2′), 6.34 (dq, J = 15.6, 1.1 Hz, 1H, H-1″), 6.11 (dq, J = 15.6, 6.8 Hz, 1H, H-2″), 5.13 (d, J = 9.2 Hz, 1H, H-2), 3.88 (s, 3H, OMe), 3.79 (s, 3H, OMe), 3.45 (m, 1H, H-3), 2.59 (q, J = 7.4 Hz, 2H, CH2CH3), 1.86 (dd, J = 6.8, 1.1 Hz, 3H, H-3″), 1.38 (d, J = 6.8 Hz, 3H, Me-C-3), 1.25 (t, J = 7.4 Hz, 3H, CH2CH3); 13C-NMR (CDCl3) δC 172.5, 151.2, 146.5, 144.1, 139.7, 139.1, 133.0, 132.4, 130.9, 123.6, 122.6, 118.7, 113.3, 110.3, 109.3, 93.1, 56.0, 45.8, 29.7, 27.3, 18.3, 17.9, 9.1; IR (KBr film) νmax 2928, 2858, 1768, 1605, 1499, 1465, 1274, 1125, 1032, 954, 822, 759 cm−1; EI-MS m/z (%) (70 eV) 382 (M+, 25; C23H26O5), 326 (100), 314 (7), 199 (6), 149 (11), 97 (16), 85 (15), 71 (22), 57 (34).
Compound 12 was prepared from 1 using esteration reaction; the reaction was similar to the preparation of 10 with butanoyl chloride and triethylamine. Compound 1 (120.4 mg) gave 12 as a colorless solid (131.2 mg, 92% yield; with solvent system EtOAc: hexane = 1:9); mp 174–175 °C; 1H-NMR (CDCl3) δH 7.03 (s, 1H, H-4), 6.95–6.97 (m, 2H, H-5′, H-6′), 6.77 (s, 1H, H-6), 6.75 (s, 1H, H-2′), 6.35 (dq, J = 15.6, 1.0 Hz, 1H, H-1″), 6.10 (dq, J = 15.6, 6.6 Hz, 1H, H-2″), 5.14 (d, J = 9.2 Hz, 1H, H-2), 3.88 (s, 3H, OMe), 3.79 (s, 3H, OMe), 3.43 (m, 1H, H-3), 2.54 (t, J = 7.3 Hz, 2H, CH2CH2CH3), 1.85 (dd, J = 6.6, 1.0 Hz, 3H, H-3″), 1.78 (sex, J = 7.3 Hz, 2H, CH2CH2CH3), 1.39 (d, J = 6.8 Hz, 3H, Me-C-3), 0.99 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (CDCl3) δC 171.5, 151.1, 146.4, 144.0, 139.6, 139.0, 132.9, 132.3, 130.8, 123.4, 122.5, 118.5, 113.2, 110.2, 109.3, 93.0, 55.8, 55.7, 45.7, 35.7, 18.4, 18.2, 17.7, 14.0; IR (KBr film) νmax 3018, 2968, 2879, 1772, 1607, 1512, 1123, 1031, 954, 826, 596, 537 cm−1; EI-MS m/z (%) (70 eV) 396 (M+, 27; C24H28O5), 326 (100), 238 (8), 178 (27), 151 (24), 140 (25), 126 (23), 85 (21), 71 (54).
Compound 13 was prepared from 1 using DDQ (dichlorodicyanobenzoquinone). Compound 1 (1.15 g, 3.50 mmol) and DDQ (0.870 g, 3.80 mmol) were dissolved in the mixture of CH2Cl: H2O = 4:1 (10 mL) and stirred for 48 h. After filtration, the product in the filtrate was purified using Si gel column chromatography. It gave compound 13 (1.05 g, 88% yield); mp 177–178°C; 1H-NMR (CDCl3) δH 9.64 (d, J = 7.6 Hz, 1H, H-3″), 7.41 (d, J = 15.8 Hz, 1H, H-1″), 7.02 (s, 1H, H-4), 6.99 (s, 1H, H-6), 6.97 (d, J = 8.1 Hz, 1H, H-6′), 6.89 (s, 1H, H-2′), 6.87 (1d, J = 8.1Hz, 1H, H-5′), 6.60 (dd, J = 15.8, 7.6 Hz, H-2″), 5.66 (s, 1H, ph-OH), 5.18 (d, J = 9.2 Hz, 1H, H-2), 3.50 (m, 1H, H-3), 1.40 (d, J = 6.8 Hz, 3H, C-3-Me); 13C-NMR (CDCl3) δC 193.6, 153.2, 150.6, 146.7, 146.0, 144.6, 134.0, 131.2, 128.1, 126.3, 119.9, 117.3, 114.3, 111.8, 108.9, 94.5, 56.0, 55.9, 45.1, 17.7; IR (KBr film) νmax 3486, 2985, 2852, 2851, 2734, 1684, 1620, 1478, 1133, 821 cm−1; EI-MS m/z (%) (70 eV) 340 (M+, 100; C20H20O5), 325 (7), 137 (15), 97 (15), 71 (18), 57 (30).
Compound 14 was prepared from 2 using SeO2 oxidative reaction. The solution of 2 (212.4 mg in 20 mL of EtOH) with SeO2 (0.14 g, 1.24 mmol) was refluxed. After 12 h, the mixture was evaporated under reduced pressure. Then, 30 mL of EtOAc was added, the mixture was filtrated by celite, and the filtrate was evaporated under reduced pressure. Then, the residue was purified by Si gel column chromatography to give 14 as a colorless solid (170.3 mg, 77% yield; with solvent system EtOAc: hexane = 1:5); mp 234–235 °C; 1H-NMR (CDCl3) δH 10.3 (s, 1H, OHC-C-3), 9.72 (d, J = 7.7 Hz, 1H, H-3″), 8.05 (d, J = 1.9 Hz, 1H, H-4), 7.58 (d, J = 15.9 Hz, 1H, H-1″), 7.41 (dd, J = 8.0, 1.8 Hz, 1H, H-6′), 7.37 (d, J = 1.9 Hz, 1H, H-6), 7.09 (d, J = 1.8 Hz, 1H, H-2′), 7.07 (d, J = 8.0 Hz, 1H, H-5′), 6.75 (dd, J = 15.9, 7.7 Hz, 1H, H-2″), 6.02 (br s, 1H, Ph-OH), 4.06 (s, 3H, OMe), 4.01 (s, 3H, OMe); 13C-NMR (CDCl3) δC 194.3, 186.6, 165.7, 153.8, 150.4, 148.2, 145.0, 143.7, 132.3, 128.4, 127.4, 123.1, 118.5, 116.2, 116.0, 115.3, 112.4, 107.0, 56.2, 55.9; IR (KBr film) νmax 3488, 2930, 2854, 2734, 1680, 1619, 1513, 1478, 1437, 1279, 1133, 1027, 822 cm−1; EI-MS m/z (%) (70 eV) 352 (M+, 100; C20H16O6), 323 (40), 296 (15), 281 (20), 253 (33), 181 (18), 165 (20), 152 (23), 105 (13), 69 (14).
Compound 15 was prepared from 14 using alkylation reaction; the reaction was similar to the preparation of 5. Compound 14 (100.5 mg) obtained 15 as a colorless solid (126.2 mg, 87% yield; with solvent system EtOAc: hexane = 1:9); mp 265–266 °C; 1H-NMR (CDCl3) δH 10.27 (s, 1H, OHC-C-3), 9.68 (d, J = 7.6 Hz, 1H, H-3″), 8.01 (s, 1H, H-4), 7.53 (d, J = 15.6 Hz, 1H, H-1″), 7.35–7.45 (m, 7H, CH2Ph, H-6, H-6′), 7.05 (s, 1H, H-2′), 7.00 (d, J = 8.0 Hz, 1H, H-5′), 6.72 (dd, J = 15.6, 7.6 Hz, 1H, H-2″), 5.22 (s, 2H, CH2Ph), 4.04 (s, 3H, OMe), 3.97 (s, 3H, OMe); 13C-NMR (CDCl3) δC 193.4, 186.3, 166.2, 152.9, 151.2, 149.9, 145.3, 144.6, 145.3, 144.6, 136.1, 132.0, 128.7, 128.3, 128.1, 127.8, 127.2, 122.9, 120.8, 116.6, 113.5, 111.8, 106.6, 70.9, 56.3, 56.1; IR (KBr film) νmax 3060, 2947, 2841, 2739, 1680, 1607, 1517, 1477, 1271, 1126, 1028, 970, 730 cm−1; EI-MS m/z (%) (70 eV) 442 (M+, 53; C27H22O6), 351 (54), 105 (11), 91 (100), 65 (5).
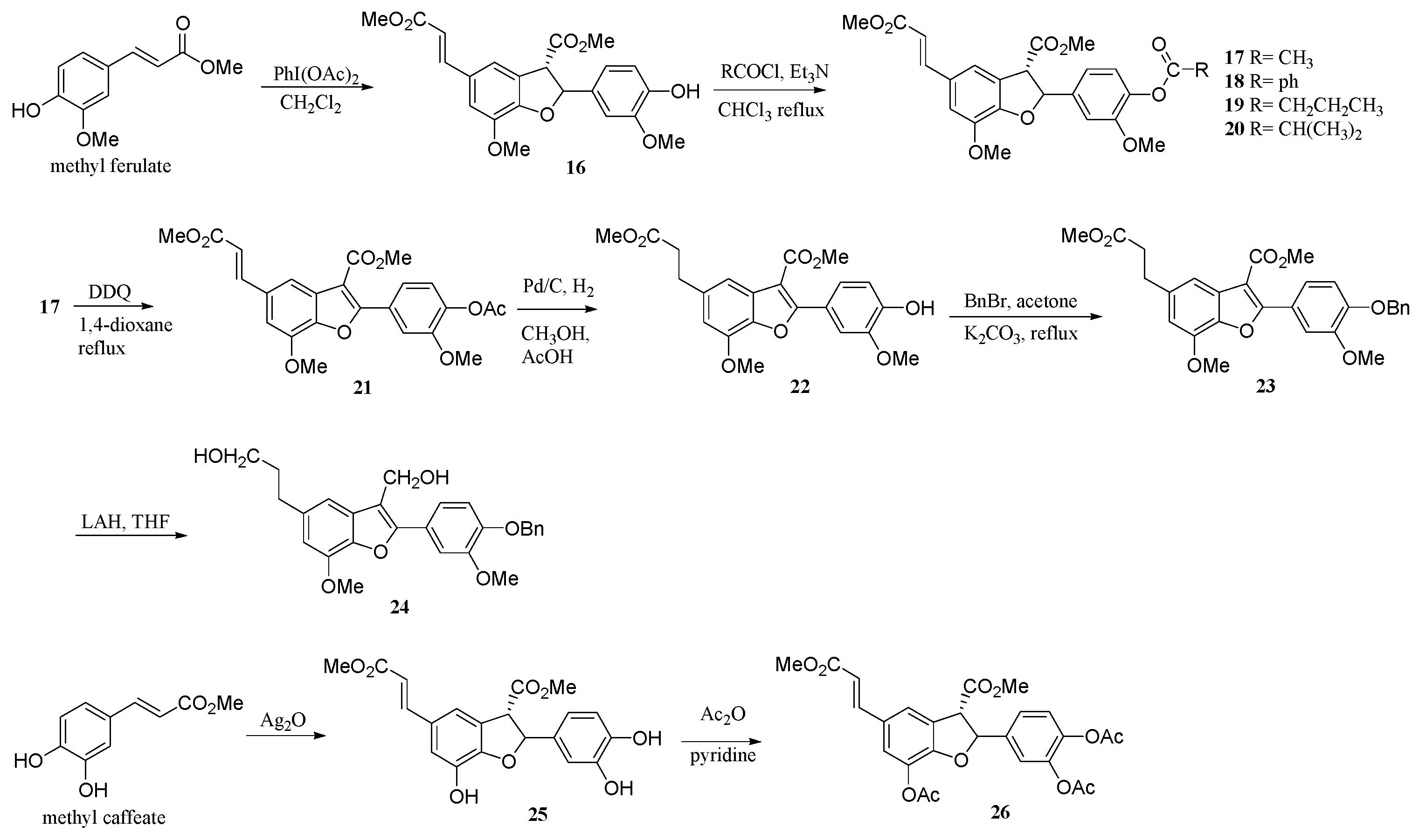
Compound 16 was prepared from methyl ferulate using IDA; the procedure was similar to the preparation of 1 from isoeugenol. Methyl ferulate (10.0 g) yielded 16 as a colorless solid (7.3 g, 73% yield; with solvent system EtOAc: hexane = 1:9 on open column); mp 96–97 °C; 1H-NMR (CDCl3) δH 7.64 (d, J = 15.9 Hz, 1H, H-1″), 7.17 (s, 1H, H-4), 7.00 (s, 1H, H-6), 6.88 (s, 3H, H-2′, H-5′, H-6′), 6.30 (d, J = 15.9 Hz, 1H, H-2″), 6.09 (d, J = 8.1 Hz, 1H, H-2), 5.63 (s, 1H, Ph-OH), 4.33 (d, J = 8.1 Hz, 1H, H-3), 3.89 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.81 (s, 3H, COOMe), 3.79 (s, 3H, COOMe); 13C-NMR (CDCl3) δC 170.7, 167.6, 146.0, 144.7, 144.6, 131.3, 128.5, 125.6, 119.4, 117.8, 115.5, 114.5, 112.0, 108.7, 87.4, 56.0, 55.9, 55.4, 52.8, 51.6; IR (KBr film) νmax 3396, 3011, 2956, 2849, 1741, 1637, 1606, 1496, 1440, 1287, 837, 612 cm−1; EI-MS m/z (%) (70 eV) 414 (M+, 95; C22H22O8), 382 (100), 350 (73), 280 (15), 266 (12), 167 (8), 151 (7), 137 (6), 58 (18).
Compound 17 was prepared from 16 using ethanoyl chloride in triethylamine; the reaction was similar to the preparation of 10. Compound 16 (1.36 g) could give 17 as a colorless solid (1.40 g, 91% yield; with solvent system EtOAc: hexane = 1:9); mp 98–99 °C; 1H-NMR (CDCl3) δH 7.63 (d, J = 15.8 Hz, 1H, H-1″), 7.17 (s, 1H, H-4), 6.97–7.00 (m, 3H, H-6, H-5′, H-6′), 6.30 (d, J = 15.8 Hz, 1H, H-2″), 6.16 (d, J = 8.0 Hz, 1H, H-2), 4.32 (d, J = 8.0 Hz, 1H, H-3), 3.91 (s, 3H, OMe), 3.83 (s, 3H, OMe), 3.80 (s, 3H, CO2Me), 3.79 (s, 3H, CO2Me), 2.29 (s, 3H, COMe); 13C-NMR (CDCl3) δC 170.5, 168.8, 167.5, 151.2, 149.7, 144.6, 144.5, 139.7, 138.4, 128.7, 125.3, 122.9, 118.1, 117.8, 115.6, 112.1, 109.9, 86.6, 56.0, 55.8, 55.4, 52.8, 51.5, 20.5; IR (KBr film) νmax 3073, 2955, 2848, 1769, 1742, 1710, 1638, 1604, 1499, 1443, 1275, 1196, 1150, 835 cm−1; EI-MS m/z (%) (70 eV) 456 (M+, 2; C24H24O9), 412 (4), 400 (5), 310 (7), 250 (6), 204 (10), 148 (100), 131 (51), 130 (17), 97 (17), 69 (23), 57 (31).
Compound 18 was prepared from 16 using benzoyl chloride and triethylamine; the reaction was similar to the preparation of 10. Compound 16 (74.1 mg) afforded 18 as a colorless solid (87.1 mg, 94% yield; with solvent system EtOAc: hexane = 1:9); mp 143–144 °C; 1H-NMR (CDCl3) δH 8.19 (d, J = 7.5 Hz, 2H, -COPh (o)), 7.64 (d, J = 15.6 Hz, 1H, H-1″), 7.59 (t, J = 7.5 Hz, 1H, -COPh (p)), 7.48 (t, J = 7.5 Hz, 2H, -COPh (m)), 7.19 (s, 1H, H-4), 7.13 (d, J = 8.0 Hz, 1H, H-5′), 7.00–7.05 (m, 4H, H-6, H-2′, H-6′), 6.67 (d, J = 15.6 Hz, 1H, H-2″), 6.20 (d, J = 8.0 Hz, 1H, H-2), 4.36 (d, J = 8.0 Hz, 1H, H-3), 3.92 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.79 (s, 6H, CO2Me); 13C-NMR (CDCl3) δC 170.6, 167.6, 164.6, 151.6, 149.8, 144.6, 140.1, 138.5, 133.5, 130.3, 129.2, 128.8, 128.5, 125.5, 123.2, 118.2, 117.9, 115.7, 112.2, 110.2, 86.8, 56.2, 56.0, 55.6, 52.9, 51.6; IR (KBr film) νmax 3069, 2950, 2852, 1734, 1641, 1607, 1506, 1446, 1274, 1209, 1028, 846, 704 cm−1; EI-MS m/z (%) (70 eV) 518 (M+, 35; C29H26O9), 486 (2), 455 (3), 382 (2), 264 (3), 220 (3), 160 (6), 105 (100), 77 (21), 57 (7).
Compound
19 was prepared from
16 using butanoyl chloride in triethylamine; the reaction was similar to the preparation of
10. Compound
16 (91.6 mg) gave
19 as a colorless solid (96.4 mg, 90% yield; with solvent system EtOAc: hexane = 1:9); mp 155–156 °C;
1H-NMR (CDCl
3) δ
H 7.62 (d, J = 15.8 Hz, 1H, H-1″), 7.17 (s, 1H, H-4), 6.97–7.01 (m, 3H, H-6, H-5′, H-6′), 6.30 (d, J = 15.8 Hz, 1H, H-2″), 6.16 (d, J = 8.0 Hz, 1H, H-2), 4.32 (d, J = 8.0 Hz, 1H, H-3), 3.91 (s, 3H, OMe), 3.83 (s, 3H, OMe), 3.78 (s, 6H, CO
2Me), 2.53 (t, J = 7.3 Hz, 2H, C
H2CH
2CH
3), 1.75 (sex, J = 7.3 Hz, 2H, CH
2C
H2CH
3), 1.02 (t, J = 7.3 Hz, 3H, CH
2CH
2C
H3);
13C-NMR (CDCl
3) δ
C 171.6, 170.6, 167.6, 151.4, 149.8, 144.7, 144.6, 140.0, 138.3, 128.8, 125.5, 123.1, 118.2, 117.9, 115.7, 112.2, 110.1, 86.8, 56.2, 55.9, 55.5, 52.9, 51.6, 35.8, 18.5, 13.5; IR (KBr film) ν
max 3003, 2960, 2850, 1766, 1742, 1711, 1608, 1507, 1434, 1275, 1141, 1030, 843 cm
−1; EI-MS
m/
z (%) (70 eV) 484 (M
+, 41; C
26H
28O
9), 414 (38), 382 (100), 350 (33), 323 (10), 290 (7), 166 (7), 71 (17) (
Figures S5–S8).
Compound
20 was prepared from
16 using isobutanoyl chloride in triethylamine; the reaction was similar to the preparation of
10. Compound
16 (91.6 mg) obtained
20 as a colorless solid (85.6 mg, 88% yield; with solvent system EtOAc: hexane = 1:9); mp 132–133 °C;
1H-NMR (CDCl
3) δ
H 7.63 (d, J = 15.8 Hz, 1H, H-1″), 7.17 (s, 1H, H-4), 6.96–6.98 (m, 4H, H-2′, H-5′, H-6′, H-6), 6.30 (d, J = 15.8 Hz, 1H, H-2″), 6.16 (d, J = 8.0 Hz, 1H, H-2), 4.32 (d, J = 8.0 Hz, 1H, H-3), 3.95 (s, 3H, OMe), 3.83 (s, 3H, OMe), 3.76 (s, 3H, CO
2Me), 3.74 (s, 3H, CO
2Me), 2.81 (m, 1H, C
H(CH
3)
2), 1.27 (d, J = 7.1 Hz, 6H, CH(C
H3)
2);
13C-NMR (CDCl
3) δ
C 175.1, 170.6, 167.5, 151.4, 149.8, 144.7, 144.6, 140.1, 138.2, 128.8, 125.5, 123.0, 118.2, 117.9, 115.7, 112.2, 110.1, 86.8, 56.1, 56.0, 55.5, 52.9, 51.6, 33.9, 18.9; IR (KBr film) ν
max 2952, 2849, 1762, 1605, 1508, 1464, 1281, 848, 757 cm
−1; EI-MS
m/
z (%) (70 eV) 484 (M
+, 45; C
26H
28O
9), 415 (27), 382 (100), 350 (28), 290 (5), 235 (7), 167 (4), 71 (13) (
Figures S9–S13).
Compound 21 was prepared from 17 using DDQ (dichlorodicyanobenzoquinone) dehydrogenative and oxidative coupling reaction; the reaction was similar to the preparation of 2. Compound 17 (1.10 g) afforded 21 as a colorless solid (0.93 g, 85% yield; with solvent system EtOAc: hexane = 1:5); mp 176–177 °C; 1H-NMR (CDCl3) δH 7.81 (s, 1H, H-2′), 7.79 (d, J = 16.0 Hz, 1H, H-1″), 7.78 (d, J = 1.3 Hz, 1H, H-4), 7.65 (dd, J = 8.3, 2.0 Hz, 1H, H-6′), 7.13 (d, J = 8.3 Hz, 1H, H-5′), 7.01 (d, J = 1.3 Hz, 1H, H-6), 6.44 (d, J = 16.0 Hz, 1H, H-2″), 4.03 (s, 3H, OMe), 3.95 (s, 3H, OMe), 3.91 (s, 3H, CO2Me), 3.81 (s, 3H, CO2Me), 2.33 (s, 3H, OAc); 13C-NMR (CDCl3) δC 168.6, 167.4, 163.9, 160.5, 150.7, 145.3, 145.2, 141.5, 131.5, 129.0, 127.6, 122.6, 122.3, 117.1, 116.1, 113.8, 109.2, 106.0, 56.1, 56.0, 51.8, 51.7, 20.6; IR (KBr film) νmax 3096, 2951, 2846, 1769, 1721, 1633, 1605, 1468, 1258, 1045, 885, 855, 597 cm−1; EI-MS m/z (%) (70 eV) 454 (M+, 10; C24H22O9), 412 (100), 382 (15), 349 (8), 323 (2), 228 (2), 151 (2), 91 (2), 69 (3).
Compound 22 was prepared from 21 using hydrogenation reductive reaction. The solution of 21 (50.6 mg in 10 mL of CH3OH and 1 mL of CH3COOH) with 10% Pd/C (10 mg) was stirred under H2 at room temperature. After 6 h, the mixture was filtrated. After removing acid with aqueous NaHCO3, the product residue was purified by Si gel column chromatography to give 22 as a colorless solid (43.8 mg, 95% yield; with solvent system EtOAc: hexane = 1:5); mp 134–135 °C; 1H-NMR (CDCl3) δH 7.67 (d, J = 1.7 Hz, 1H, H-2′), 7.59 (dd, J = 8.6, 1.7 Hz, 1H, H-6′), 7.41 (d, J = 1.1 Hz, 1H, H-4), 6.98 (d, J = 8.6 Hz, 1H, H-5′), 6.68 (d, J = 1.1 Hz, 1H, H-6), 5.88 (s, 1H, Ph-OH), 3.99 (s, 3H, OMe), 3.96 (s, 3H, OMe), 3.92 (s, 3H, CO2Me), 3.68 (s, 3H, CO2Me), 3.04 (t, J = 7.5 Hz, 2H, H-1″), 2.69 (t, J = 7.5 Hz, 1H, H-2″); 13C-NMR (CDCl3) δC 173.4, 164.6, 147.7, 145.9, 144.7, 141.6, 137.3, 129.0, 123.7, 121.5, 114.1, 113.7, 112.2, 107.8, 56.1, 56.0, 51.6, 51.5, 34.6, 31.5; IR (KBr film) νmax 3424, 2954, 2855, 1714, 1602, 1514, 1449, 1276, 1209, 1046, 829, 787 cm−1; EI-MS m/z (%) (70 eV) 414 (M+, 100; C22H22O8), 383 (9), 354 (10), 341 (25), 323 (47), 170 (5), 161 (3), 151 (2).
Compound 23 was prepared from 22 using alkylation reaction; the reaction was similar to the preparation of 9. Compound 22 (150.4 mg) afforded 23 as a colorless solid (146.4 mg, 80% yield; with solvent system EtOAc: hexane = 1:9); mp 78–79 °C; 1H-NMR (CDCl3) δH 7.67 (d, J = 2.1 Hz, 1H, H-2′), 7.57 (dd, J = 8.7, 2.1 Hz, 1H, H-6′), 7.28–7.44 (m, 6H, OCH2Ph, H-4), 6.93 (d, J = 8.7 Hz, 1H, H-5′), 6.67 (d, J = 1.2 Hz, 1H, H-6), 5.21 (s, 2H, OCH2Ph), 3.98 (s, 3H, OMe), 3.95 (s, 3H, OMe), 3.91 (s, 3H, CO2Me), 3.67 (s, 3H, OMe-C-3), 3.04 (t, J = 8.1 Hz, 2H, H-1″), 2.68 (t, J = 8.1 Hz, 1H, H-2″); 13C-NMR (CDCl3) δC 173.3, 164.6, 161.1, 150.0, 148.9, 144.7, 141.7, 137.4, 136.7, 129.0, 128.6, 127.9, 127.2, 122.9, 122.4, 113.7, 113.1, 112.9, 108.0, 107.9, 70.8, 56.2, 56.1, 51.6, 51.5, 36.4, 31.6; IR (KBr film) νmax 2933, 2857, 1737, 1717, 1603, 1511, 1265, 1236, 1147, 1096, 1048, 742 cm−1; EI-MS m/z (%) (70 eV) 504 (M+, 28; C29H28O8), 413 (100), 382 (7), 353 (5), 341 (7), 323 (11), 91 (45).
Compound 24 was prepared from 23 using lithium aluminum hydride (LAH) reduction reaction. The solution of 23 (122.3 mg, in 20 mL of dry THF) was cooled to –10 °C before LAH (150.3 mg) was added. The solution was stirred under –10 °C. After 8 h, wet THF (10 mL) was added to the solution dropwise to quench the reaction. The solution was adjusted to pH = 4 using HCl (3N), and then the solution was evaporated under reduced pressure. The solution was extracted by EtOAc (3 × 100 mL). The combined organic layer was washed with saturated NaHCO3 solution and brine and dry (Na2SO4), and the solvent was removed under reduced pressure to give a residue. Then, the residue was purified using Si gel column chromatography to give 24 as a colorless solid (103.2 mg, 95% yield; with solvent system EtOAc: hexane = 1:4); mp 187–188 °C; 1H-NMR (CDCl3) δH 7.29–7.45 (m, 7H, OCH2Ph, H-2′, H-6′), 7.07 (s, 1H, H-4), 6.95 (d, J = 8.3 Hz, 1H, H-5′), 6.65 (s, 1H, H-6), 5.20 (s, 2H, OCH2Ph), 4.88 (s, 2H, HOCH2-C-3), 3.98 (s, 3H, OMe), 3.96 (s, 3H, OMe), 3.70 (t, J = 7.2 Hz, 2H, H-3″), 2.79 (t, J = 7.2 Hz, 2H, H-1″), 1.94 (quin, J = 7.2 Hz, 2H, H-2″); 13C-NMR (CDCl3) δC 154.2, 149.7, 149.6, 148.9, 144.8, 141.6, 137.7, 137.6, 136.8, 136.7, 136.6, 131.1, 128.6, 127.9, 127.3, 127.2, 123.4, 120.4, 113.9, 113.8, 113.6, 111.0, 110.8, 107.7, 70.9, 62.2, 60.4, 56.2, 56.1, 34.6, 32.4; IR (KBr film) νmax 2933, 2857, 1737, 1717, 1603, 1511, 1265, 1236, 1147, 1096, 1048, 742 cm−1; HR-ESI-MS (M+H)+ m/z 449.1954; (C27H29O6).
Preparation of
25: Methyl caffeate (762.2 mg) was dissolved in a mixture of benzene (20 mL) and acetone (30 mL), and then Ag
2O (1.82 g) was added. The reaction mixture was stirred at room temperature for 60 h. The precipitation was removed with filtration, and the filtrate gave
25 (306.9 mg, 45% yield). Compound
25: mp 187–188 °C;
1H-NMR (CDCl
3) δ
H 7.54 (d, J = 15.9 Hz, 1H, H-1″), 7.05 (s, 1H, H-4), 6.99 (s, 1H, H-6), 6.85 (d, J = 1.9 Hz, 1H, H-2′), 6.82 (d, J = 8.1 Hz, H-5′), 6.76 (dd, J = 8.1, 1.9 Hz, 1H, H-6′), 6.24 (d, J = 15.9 Hz, 1H, H-2″), 6.02 (d, 2H, J = 7.4 Hz, 1H, H-2), 4.26 (d, J = 7.4 Hz, 1H, H-3), 3.79 (s, 3H, -OC
H3), 3.77 (s, 3H, OC
H3);
13C-NMR (CDCl
3) δ
C 171.0, 168.3, 148.5, 145.0, 144.7, 144.4, 144.0, 140.4, 132.0, 128.7, 125.4, 118.7, 117.6, 115.5, 115.4, 113.0, 87.2, 55.6, 53.0, 51.8; IR (KBr film) ν
max 3397, 2958, 1739, 1697, 1609, 1506, 1444, 1281, 1198, 980, 854, 814 cm
−1; EI-MS
m/
z (%) (70 eV) 386 (M
+, 37; C
20H
18O
8), 354 (35), 322 (100), 294 (27), 267 (13), 194 (55), 163 (52), 134 (14) (
Figures S13–S16).
Compound 26 was prepared from 25 (510.7 mg) under usual acetylation conditions using Ac2O and pyridine. Compound 26 (609.6 mg, 90% yield), mp 137–139 °C; 1H-NMR (CDCl3) δH 7.59 (d, J = 15.8 Hz, 1H, H-1″), 7.42 (s, 1H, H-4), 7.28 (dd, J = 8.4, 1.9 Hz, 1H, H-6′), 7.21 (d, J = 1.9 Hz, 1H, H-2′), 7.19 (s, 1H, H-6), 7.17 (d, J = 8.4 Hz, 1H, H-5′), 6.29 (d, J = 15.8 Hz, 1H, H-2″), 6.19 (d, J = 7.4 Hz, 1H, H-2), 4.28 (d, J=7.4 Hz, 1H, H-3), 3.83 and 3.78 (s each, 3H-OCH3), 2.30 (s, 3H, OAc), and 2.27 (s, 6H, OAc); IR (KBr film) νmax 3074, 3016, 1776, 1739, 1716, 1643, 1612, 1591, 1273, 1203, 1176 cm−1; HR-ESI-MS ((M+H)+ m/z 513.4708; C26H25O11).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}