
Targeted Therapies in Psoriatic Arthritis—An Update

Abstract
:1. Introduction
2. Immunopathology of PsA, Genetics, and Environment
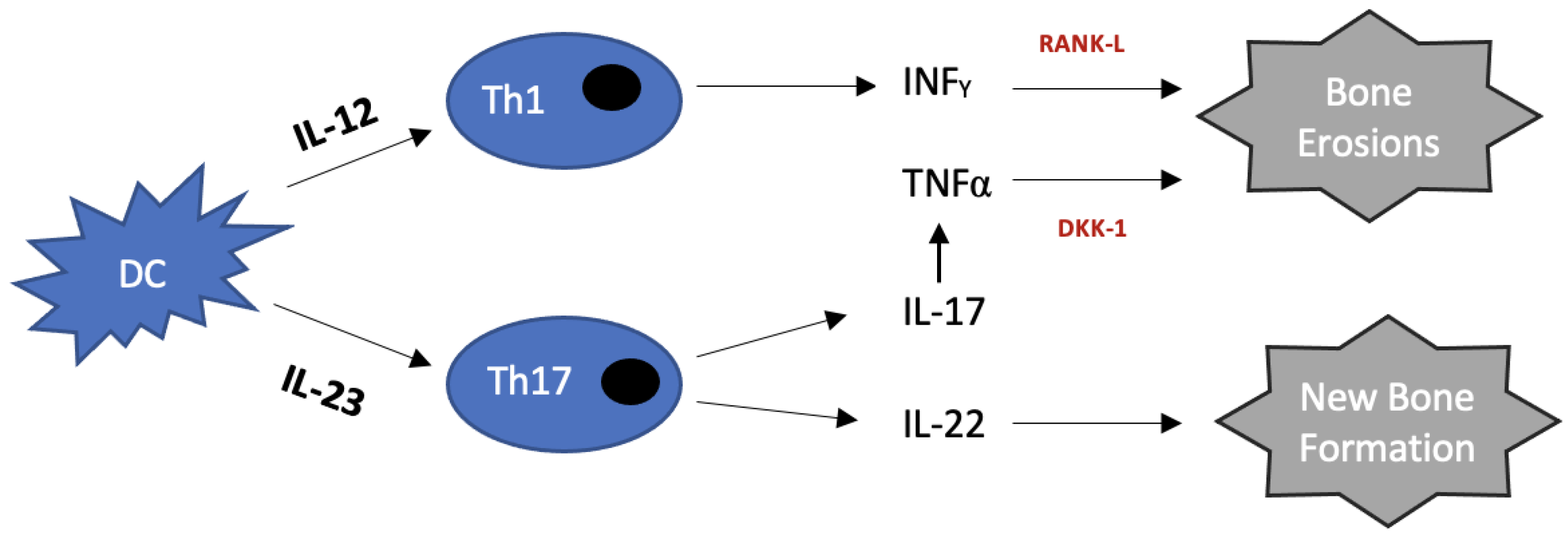
2.1. Immunopathology
2.2. Genetics
2.3. Environment
3. Unmet Needs in Therapeutics—The Challenges
4. TNFα
5. IL-12/23
6. IL-23
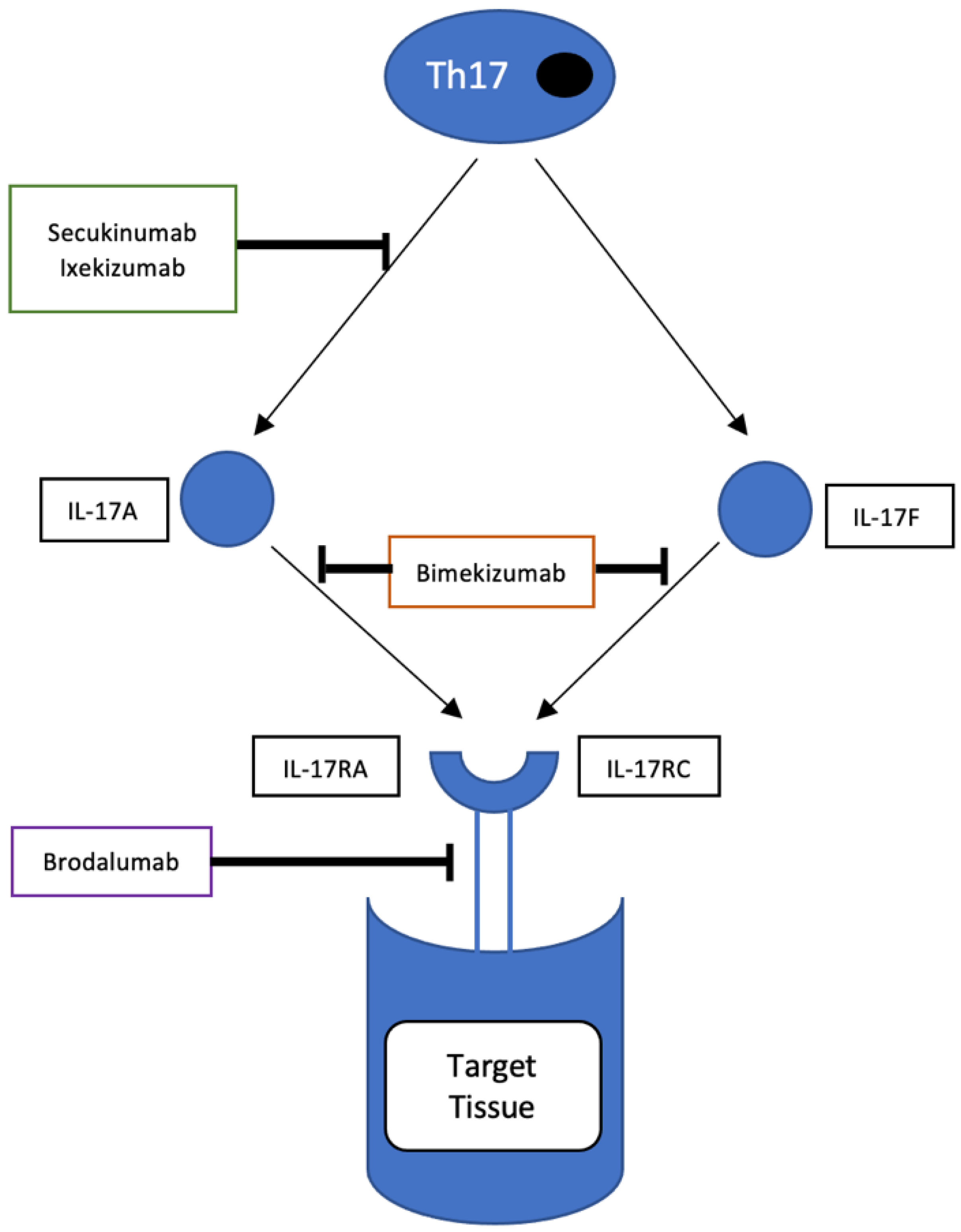
7. IL17
8. Janus Kinase (JAK) Inhibitors
9. Other Targets
10. Future Direction and Remaining Questions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alinaghi, F.; Calov, M.; Kristensen, L.E.; Gladman, D.D.; Coates, L.C.; Jullien, D.; Gottlieb, A.B.; Gisondi, P.; Wu, J.J.; Thyssen, J.P.; et al. Prevalence of psoriatic arthritis in patients with psoriasis: A systematic review and meta-analysis of observational and clinical studies. J. Am. Acad. Dermatol. 2019, 80, 251–265.e219. [Google Scholar] [CrossRef] [Green Version]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and incidence of psoriatic arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Qi, F.; Tan, Y.; Yao, A.; Yang, X.; He, Y. Psoriasis to Psoriatic Arthritis: The Application of Proteomics Technologies. Front. Med. 2021, 8, 681172. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, D.; Stafford, L.; Bresnihan, B.; FitzGerald, O. A prospective, clinical and radiological study of early psoriatic arthritis: An early synovitis clinic experience. Rheumatology 2003, 42, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Haroon, M.; Gallagher, P.; FitzGerald, O. Diagnostic delay of more than 6 months contributes to poor radiographic and functional outcome in psoriatic arthritis. Ann. Rheum. Dis. 2015, 74, 1045. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Helliwell, P.; Ritchlin, C.T. Psoriatic Arthritis and Burden of Disease: Patient Perspectives from the Population-Based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) Survey. Rheumatol. Ther. 2016, 3, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, B.C.K.; Jadon, D.R. Unmet needs in psoriatic arthritis. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101693. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Barnes, L.; Rogers, S.; FitzGerald, O. Immunohistochemical markers for arthritis in psoriasis. Ann. Rheum. Dis. 1994, 53, 450. [Google Scholar] [CrossRef] [Green Version]
- Veale, D.; Yanni, G.; Rogers, S.; Barnes, L.; Bresnihan, B.; Fitzgerald, O. Reduced synovial membrane macrophage numbers, elam-1 expression, and lining layer hyperplasia in psoriatic arthritis as compared with rheumatoid arthritis. Arthritis Rheum. 1993, 36, 893–900. [Google Scholar] [CrossRef]
- Van Kuijk, A.W.R.; Reinders-Blankert, P.; Smeets, T.J.M.; Dijkmans, B.A.C.; Tak, P.P. Detailed analysis of the cell infiltrate and the expression of mediators of synovial inflammation and joint destruction in the synovium of patients with psoriatic arthritis: Implications for treatment. Ann. Rheum. Dis. 2006, 65, 1551. [Google Scholar] [CrossRef] [Green Version]
- Yeremenko, N.; Paramarta, J.E.; Baeten, D. The interleukin-23/interleukin-17 immune axis as a promising new target in the treatment of spondyloarthritis. Curr. Opin. Rheumatol. 2014, 26, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.C.; FitzGerald, O.; Helliwell, P.S.; Paul, C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: Is all inflammation the same? Semin. Arthritis Rheum. 2016, 46, 291–304. [Google Scholar] [CrossRef] [Green Version]
- De Vlam, K.; Gottlieb, A.B.; Mease, P.J. Current concepts in psoriatic arthritis: Pathogenesis and management. Acta Derm. Venereol. 2014, 94, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Lories, R.J.; McInnes, I.B. Primed for inflammation: Enthesis-resident T cells. Nat. Med. 2012, 18, 1018–1019. [Google Scholar] [CrossRef]
- Veale, D.J.; Fearon, U. What makes psoriatic and rheumatoid arthritis so different? RMD Open 2015, 1, e000025. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; Kavanaugh, A.; Genovese, M.; Ritchlin, C.; Rosengren, S.; Quistberg, A. Rituximab in psoriatic arthritis provides modest clinical improvement and reduces expression of inflammatory biomarkers in skin lesions. Arthritis Rheum. 2010, 62, S818. [Google Scholar]
- Reece, R.J.; Canete, J.D.; Parsons, W.J.; Emery, P.; Veale, D.J. Distinct vascular patterns of early synovitis in psoriatic, reactive, and rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Ritchlin, C.; FitzGerald, O. Immunopathology of psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 2005, 64, ii26. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Gibbon, W.; Emery, P. Classification of inflammatory arthritis by enthesitis. Lancet 1998, 352, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+ CD4− CD8− entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Schentag, C.T.; Brockbank, J.E.; Pellett, F.J.; Shanmugarajah, S.; Toloza, S.M.A.; Rahman, P.; Gladman, D.D. Familial aggregation of psoriatic arthritis. Ann. Rheum. Dis. 2009, 68, 664. [Google Scholar] [CrossRef]
- Haroon, M.; Winchester, R.; Giles, J.T.; Heffernan, E.; FitzGerald, O. Certain class I HLA alleles and haplotypes implicated in susceptibility play a role in determining specific features of the psoriatic arthritis phenotype. Ann. Rheum. Dis. 2016, 75, 155. [Google Scholar] [CrossRef] [PubMed]
- Pattison, E.; Harrison, B.J.; Griffiths, C.E.M.; Silman, A.J.; Bruce, I.N. Environmental risk factors for the development of psoriatic arthritis: Results from a case–control study. Ann. Rheum. Dis. 2008, 67, 672. [Google Scholar] [CrossRef]
- Van den Bosch, F.; Coates, L. Clinical management of psoriatic arthritis. Lancet 2018, 391, 2285–2294. [Google Scholar] [CrossRef]
- Gossec, L.; Baraliakos, X.; Kerschbaumer, A.; de Wit, M.; McInnes, I.; Dougados, M.; Primdahl, J.; McGonagle, D.G.; Aletaha, D.; Balanescu, A.; et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann. Rheum. Dis. 2020, 79, 700. [Google Scholar] [CrossRef]
- Coates, L.C.; Soriano, E.R.; Corp, N.; Bertheussen, H.; Callis Duffin, K.; Campanholo, C.B.; Chau, J.; Eder, L.; Fernández-Ávila, D.G.; FitzGerald, O.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): Updated treatment recommendations for psoriatic arthritis 2021. Nat. Rev. Rheumatol. 2022, 18, 465–479. [Google Scholar] [CrossRef]
- Genovese, M.C.; Weinblatt, M.E.; Mease, P.J.; Aelion, J.A.; Peloso, P.M.; Chen, K.; Li, Y.; Liu, J.; Othman, A.A.; Khatri, A.; et al. Dual inhibition of tumour necrosis factor and interleukin-17A with ABT-122: Open-label long-term extension studies in rheumatoid arthritis or psoriatic arthritis. Rheumatology 2018, 57, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.J.; Genovese, M.C.; Weinblatt, M.E.; Peloso, P.M.; Chen, K.; Othman, A.A.; Li, Y.; Mansikka, H.T.; Khatri, A.; Wishart, N.; et al. Phase II Study of ABT-122, a Tumor Necrosis Factor- and Interleukin-17A-Targeted Dual Variable Domain Immunoglobulin, in Patients With Psoriatic Arthritis With an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2018, 70, 1778–1789. [Google Scholar] [CrossRef] [Green Version]
- Thibodeaux, Q.; Ly, K.; Reddy, V.; Smith, M.P.; Liao, W. Dual biologic therapy for recalcitrant psoriasis and psoriatic arthritis. JAAD Case Rep. 2019, 5, 928–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Research, J.; Development, L. A Study of Guselkumab and Golimumab Combination Therapy in Participants with Active Psoriatic Arthritis. 2021. Available online: https://ClinicalTrials.gov/show/NCT05071664 (accessed on 13 December 2022).
- Zardin-Moraes, M.; da Silva, A.L.F.A.; Saldanha, C.; Kohem, C.L.; Coates, L.C.; Henrique, L.R.; Palominos, P.E.; Chakr, R.M.d.S. Prevalence of Psoriatic Arthritis Patients Achieving Minimal Disease Activity in Real-world Studies and Randomized Clinical Trials: Systematic Review with Metaanalysis. J. Rheumatol. 2020, 47, 839. [Google Scholar] [CrossRef]
- Mease, P.J.; Gladman, D.D.; Ritchlin, C.T.; Ruderman, E.M.; Steinfeld, S.D.; Choy, E.H.; Sharp, J.T.; Ory, P.A.; Perdok, R.J.; Weinberg, M.A. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: Results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005, 52, 3279–3289. [Google Scholar] [CrossRef]
- Mease, P.J.; Kivitz, A.J.; Burch, F.X.; Siegel, E.L.; Cohen, S.B.; Ory, P.; Salonen, D.; Rubenstein, J.; Sharp, J.T.; Tsuji, W. Etanercept treatment of psoriatic arthritis: Safety, efficacy, and effect on disease progression. Arthritis Rheum. 2004, 50, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Krueger, G.G.; de Vlam, K.; Birbara, C.; Beutler, A.; Guzzo, C.; Zhou, B.; Dooley, L.T.; Kavanaugh, A. Infliximab improves signs and symptoms of psoriatic arthritis: Results of the IMPACT 2 trial. Ann. Rheum. Dis. 2005, 64, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, A.; McInnes, I.; Mease, P.; Krueger, G.G.; Gladman, D.; Gomez-Reino, J.; Papp, K.; Zrubek, J.; Mudivarthy, S.; Mack, M. Golimumab, a new human tumor necrosis factor α antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four–week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum. 2009, 60, 976–986. [Google Scholar] [CrossRef]
- Mease, P.J.; Fleischmann, R.; Deodhar, A.A.; Wollenhaupt, J.; Khraishi, M.; Kielar, D.; Woltering, F.; Stach, C.; Hoepken, B.; Arledge, T.; et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a Phase 3 double-blind randomised placebo-controlled study (RAPID-PsA). Ann. Rheum. Dis. 2014, 73, 48. [Google Scholar] [CrossRef]
- Mease, P.J.; Gladman, D.D.; Collier, D.H.; Ritchlin, C.T.; Helliwell, P.S.; Liu, L.; Kricorian, G.; Chung, J.B. Etanercept and Methotrexate as Monotherapy or in Combination for Psoriatic Arthritis: Primary Results From a Randomized, Controlled Phase III Trial. Arthritis Rheumatol. 2019, 71, 1112–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.; van der Horst-Bruinsma, I.E.; Huang, F.; Burgos-Vargas, R.; Vlahos, B.; Koenig, A.S.; Freundlich, B. Clinical efficacy and safety of etanercept versus sulfasalazine in patients with ankylosing spondylitis: A randomized, double-blind trial. Arthritis Rheum. 2011, 63, 1543–1551. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Kivitz, A.; Schiff, M.H.; Sieper, J.; Dijkmans, B.A.; Braun, J.; Dougados, M.; Reveille, J.D.; Wong, R.L.; Kupper, H. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: Results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006, 54, 2136–2146. [Google Scholar] [CrossRef]
- Inman, R.D.; Davis, J.C., Jr.; Heijde, D.V.D.; Diekman, L.; Sieper, J.; Kim, S.I.; Mack, M.; Han, J.; Visvanathan, S.; Xu, Z. Efficacy and safety of golimumab in patients with ankylosing spondylitis: Results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum. 2008, 58, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Dijkmans, B.; Geusens, P.; Sieper, J.; DeWoody, K.; Williamson, P.; Braun, J. Efficacy and safety of infliximab in patients with ankylosing spondylitis: Results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum. 2005, 52, 582–591. [Google Scholar] [CrossRef]
- Landewé, R.; Braun, J.; Deodhar, A.; Dougados, M.; Maksymowych, W.; Mease, P.; Reveille, J.; Rudwaleit, M.; Van Der Heijde, D.; Stach, C. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann. Rheum. Dis. 2014, 73, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, C.; Veale, D.J. Therapeutic targets in psoriatic arthritis. Int. J. Clin. Rheumatol. 2015, 10, 489–499. [Google Scholar] [CrossRef]
- Schett, G. Bone Formation in Psoriatic Arthritis: A Report from the GRAPPA 2013 Annual Meeting. J. Rheumatol. 2014, 41, 1218. [Google Scholar] [CrossRef] [PubMed]
- Paine, A.; Ritchlin, C. Bone remodeling in psoriasis and psoriatic arthritis: An update. Curr. Opin. Rheumatol. 2016, 28, 66–75. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Haas-Smith, S.A.; Li, P.; Hicks, D.G.; Schwarz, E.M. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J. Clin. Investig. 2003, 111, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Lories, R.J.; Schett, G. Pathophysiology of new bone formation and ankylosis in spondyloarthritis. Rheum. Dis. Clin. 2012, 38, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Mantravadi, S.; Ogdie, A.; Kraft, W.K. Tumor necrosis factor inhibitors in psoriatic arthritis. Expert Rev. Clin. Pharmacol. 2017, 10, 899–910. [Google Scholar] [CrossRef]
- Johnsson, H.J.; McInnes, I.B. Interleukin-12 and interleukin-23 inhibition in psoriatic arthritis. Clin. Exp. Rheumatol. 2015, 33, S115–S118. [Google Scholar]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.C.; Wang, Y.; Li, S.; et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; McInnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.-K.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann. Rheum. Dis. 2014, 73, 990. [Google Scholar] [CrossRef] [PubMed]
- Orr, C.; Veale, D.J. Is there a need for new agents with novel mechanisms of action in psoriatic arthritis? Ann. Rheum. Dis. 2014, 73, 951. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijde, D.; Sharp, J.; Wassenberg, S.; Gladman, D.D. Psoriatic arthritis imaging: A review of scoring methods. Ann. Rheum. Dis. 2005, 64, ii61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poddubnyy, D.; Hermann, K.-G.A.; Callhoff, J.; Listing, J.; Sieper, J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: Results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann. Rheum. Dis. 2014, 73, 817–823. [Google Scholar] [CrossRef]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro-and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef]
- Sherlock, J.P.; Cua, D.J. Interleukin-23 in perspective. Rheumatology 2021, 60, iv1–iv3. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.-P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.-K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo-and active comparator–controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.-K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo-and active comparator–controlled voyage 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [PubMed] [Green Version]
- Langley, R.; Tsai, T.F.; Flavin, S.; Song, M.; Randazzo, B.; Wasfi, Y.; Jiang, J.; Li, S.; Puig, L. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: Results of the randomized, double-blind, phase III NAVIGATE trial. Br. J. Dermatol. 2018, 178, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Deodhar, A.; Helliwell, P.S.; Boehncke, W.-H.; Kollmeier, A.P.; Hsia, E.C.; Subramanian, R.A.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Rahman, P.; Gottlieb, A.B.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; Zhuang, Y. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1126–1136. [Google Scholar] [CrossRef]
- McInnes, I.B.; Rahman, P.; Gottlieb, A.B.; Hsia, E.C.; Kollmeier, A.P.; Chakravarty, S.D.; Xu, X.L.; Subramanian, R.A.; Agarwal, P.; Sheng, S. Efficacy and safety of guselkumab, an interleukin-23p19–specific monoclonal antibody, through one year in biologic-naive patients with psoriatic arthritis. Arthritis Rheumatol. 2021, 73, 604–616. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Helliwell, P.S.; Boehncke, W.-H.; Soriano, E.R.; Hsia, E.C.; Kollmeier, A.P.; Chakravarty, S.D.; Zazzetti, F.; Subramanian, R.A.; Xu, X.L. Guselkumab, an inhibitor of the IL-23p19 subunit, provides sustained improvement in signs and symptoms of active psoriatic arthritis: 1 year results of a phase III randomised study of patients who were biologic-naïve or TNFα inhibitor-experienced. RMD Open 2021, 7, e001457. [Google Scholar] [CrossRef]
- Rahman, P.; Ritchlin, C.T.; Helliwell, P.S.; Boehncke, W.-H.; Mease, P.J.; Gottlieb, A.B.; Kafka, S.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; et al. Pooled Safety Results Through 1 Year of 2 Phase III Trials of Guselkumab in Patients With Psoriatic Arthritis. J. Rheumatol. 2021, 48, 1815–1823. [Google Scholar] [CrossRef]
- McGonagle, D.; McInnes, I.B.; Deodhar, A.; Schett, G.; Shawi, M.; Kafka, S.; Karyekar, C.S.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; et al. Resolution of enthesitis by guselkumab and relationships to disease burden: 1-year results of two phase 3 psoriatic arthritis studies. Rheumatology 2021, 60, 5337–5350. [Google Scholar] [CrossRef]
- Coates, L.C.; Gossec, L.; Theander, E.; Bergmans, P.; Neuhold, M.; Karyekar, C.S.; Shawi, M.; Noël, W.; Schett, G.; McInnes, I.B. Efficacy and safety of guselkumab in patients with active psoriatic arthritis who are inadequate responders to tumour necrosis factor inhibitors: Results through one year of a phase IIIb, randomised, controlled study (COSMOS). Ann. Rheum. Dis. 2022, 81, 359. [Google Scholar] [CrossRef]
- McInnes, I.B.; Rahman, P.; Gottlieb, A.B.; Hsia, E.C.; Kollmeier, A.P.; Xu, X.L.; Jiang, Y.; Sheng, S.; Shawi, M.; Chakravarty, S.D.; et al. Long-Term Efficacy and Safety of Guselkumab, a Monoclonal Antibody Specific to the p19 Subunit of Interleukin-23, Through Two Years: Results From a Phase III, Randomized, Double-Blind, Placebo-Controlled Study Conducted in Biologic-Naive Patients With Active Psoriatic Arthritis. Arthritis Rheumatol. 2022, 74, 475–485. [Google Scholar] [CrossRef]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: Results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 2018, 77, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tok, M.N.; Na, S.; Lao, C.R.; Alvi, M.; Pots, D.; Van de Sande, M.G.; Taurog, J.D.; Sedgwick, J.D.; Baeten, D.L.; Van Duivenvoorde, L.M. The initiation, but not the persistence, of experimental spondyloarthritis is dependent on interleukin-23 signaling. Front. Immunol. 2018, 9, 1550. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; Watad, A.; Sharif, K.; Bridgewood, C. Why inhibition of IL-23 lacked efficacy in ankylosing spondylitis. Front. Immunol. 2021, 12, 614255. [Google Scholar] [CrossRef]
- Mease, P.J.; Helliwell, P.S.; Gladman, D.D.; Poddubnyy, D.; Baraliakos, X.; Chakravarty, S.D.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; et al. Efficacy of guselkumab on axial involvement in patients with active psoriatic arthritis and sacroiliitis: A post-hoc analysis of the phase 3 DISCOVER-1 and DISCOVER-2 studies. Lancet Rheumatol. 2021, 3, e715–e723. [Google Scholar] [CrossRef]
- Huynh, D.; Kavanaugh, A. Psoriatic arthritis: Current therapy and future approaches. Rheumatology 2015, 54, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 signaling: The yin and the yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, B.; Gullick, N.J.; Walter, G.J.; Rajasekhar, M.; Garrood, T.; Evans, H.G.; Taams, L.S.; Kirkham, B.W. Interleukin-17+ CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol. 2014, 66, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandus, C.; Bioley, G.; Rivals, J.P.; Dudler, J.; Speiser, D.; Romero, P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008, 58, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 2001, 194, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.J.; Ruddy, M.J.; Wong, G.C.; Sfintescu, C.; Baker, P.J.; Smith, J.B.; Evans, R.T.; Gaffen, S.L. An essential role for IL-17 in preventing pathogen-initiated bone destruction: Recruitment of neutrophils to inflamed bone requires IL-17 receptor–dependent signals. Blood 2007, 109, 3794–3802. [Google Scholar] [CrossRef]
- Thaçi, D.; Blauvelt, A.; Reich, K.; Tsai, T.F.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Hugot, S.; You, R.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J. Am. Acad. Derm. 2015, 73, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B. Secukinumab in plaque psoriasis—Results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; Van Der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; Van der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; van der Heijde, D.; Landewé, R.; Mpofu, S.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Meiser, K. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: Primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann. Rheum. Dis. 2018, 77, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, A.; McInnes, I.B.; Mease, P.J.; Hall, S.; Chinoy, H.; Kivitz, A.J.; Wang, Z.; Mpofu, S. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: Results from the randomized placebo-controlled FUTURE 2 study. J. Rheumatol. 2016, 43, 1713–1717. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Mease, P.J.; Ritchlin, C.T.; Rahman, P.; Gottlieb, A.B.; Kirkham, B.; Kajekar, R.; Delicha, E.-M.; Pricop, L.; Mpofu, S. Secukinumab sustains improvement in signs and symptoms of psoriatic arthritis: 2 year results from the phase 3 FUTURE 2 study. Rheumatology 2017, 56, 1993–2003. [Google Scholar] [CrossRef] [Green Version]
- Hueber, W.; Patel, D.D.; Dryja, T.; Wright, A.M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M.H.; Group, P.S. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; Kavanaugh, A.; Reimold, A. Sustained improvements in the signs and syptoms of active psoriatic arthritis through 3 years: Efficacy and safety results from a phase 3 trial [abstract] 2016 ACR. In Proceedings of the ARHP Annual Meeting, Washington, DC, USA, 11–16 November 2016. [Google Scholar]
- Kavanaugh, A.; Mease, P.J.; Reimold, A.M.; Tahir, H.; Rech, J.; Hall, S.; Geusens, P.; Wang, Z.; Pricop, L.; Mpofu, S. Secukinumab for long-term treatment of psoriatic arthritis: A two-year followup from a phase iii, randomized, double-blind placebo-controlled study. Arthritis Care Res. 2017, 69, 347–355. [Google Scholar] [CrossRef]
- Kavanaugh, A.; McInnes, I.B.; Hall, S.; Chinoy, H.; Kivitz, A.; Kandala, S.; Patekar, M.; Mpofu, S. THU0411 Secukinumab Efficacy in Anti-TNF-Naive and Anti-TNF-IR Patients with Psoriatic Arthritis: Results of a Phase 3 Multicenter, Double-Blind, Placebo-Controlled Study (Future 2). Ann. Rheum. Dis. 2015, 74, 345. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Mease, P.J.; Landewé, R.B.M.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Zhu, X.; Ligozio, G.; et al. Secukinumab provides sustained low rates of radiographic progression in psoriatic arthritis: 52-week results from a phase 3 study, FUTURE 5. Rheumatology 2020, 59, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Croes, M.; Öner, F.C.; van Neerven, D.; Sabir, E.; Kruyt, M.C.; Blokhuis, T.J.; Dhert, W.J.; Alblas, J. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone 2016, 84, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Wang, S.E.; Lee, Y.L.; Kang, S.; Lee, B.; Han, J.; Sung, I.-H.; Park, Y.-S.; Bae, S.-C.; Kim, T.-H. IL-17A induces osteoblast differentiation by activating JAK2/STAT3 in ankylosing spondylitis. Arthritis Res. Ther. 2018, 20, 115. [Google Scholar] [CrossRef]
- Kampylafka, E.; d’Oliveira, I.; Linz, C.; Lerchen, V.; Stemmler, F.; Simon, D.; Englbrecht, M.; Sticherling, M.; Rech, J.; Kleyer, A. Resolution of synovitis and arrest of catabolic and anabolic bone changes in patients with psoriatic arthritis by IL-17A blockade with secukinumab: Results from the prospective PSARTROS study. Arthritis Res. Ther. 2018, 20, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, P.; Kirkham, B.; Okada, M.; Rahman, P.; Combe, B.; Burmester, G.-R.; Adams, D.H.; Kerr, L.; Lee, C.; Shuler, C.L.; et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: Results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet 2017, 389, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Baraliakos, X.; Gossec, L.; Pournara, E.; Jeka, S.; Mera-Varela, A.; Angelo, S.; Schulz, B.; Rissler, M.; Nagar, K.; Perella, C.; et al. Secukinumab in patients with psoriatic arthritis and axial manifestations: Results from the double-blind, randomised, phase 3 MAXIMISE trial. Ann. Rheum. Dis. 2021, 80, 582. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Gladman, D.D.; Bolce, R.; Sandoval, D.; Park, S.Y.; Liu Leage, S.; Nash, P.; Poddubnyy, D. POS1045 Ixekizumab efficacy on spinal pain, disease activity and quality of life in patients with psoriatic arthritis presenting with symptoms suggestive of axial involvement. Ann. Rheum. Dis. 2021, 80, 798. [Google Scholar] [CrossRef]
- Mease, P.J.; Smolen, J.S.; Behrens, F.; Nash, P.; Leage, S.L.; Li, L.; Tahir, H.; Gooderham, M.; Krishnan, E.; Liu-Seifert, H. A head-to-head comparison of the efficacy and safety of ixekizumab and adalimumab in biological-naïve patients with active psoriatic arthritis: 24-week results of a randomised, open-label, blinded-assessor trial. Ann. Rheum. Dis. 2020, 79, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Behrens, F.; Mease, P.J.; Kavanaugh, A.; Ritchlin, C.; Nash, P.; Masmitja, J.G.; Goupille, P.; Korotaeva, T.; Gottlieb, A.B. Secukinumab versus adalimumab for treatment of active psoriatic arthritis (EXCEED): A double-blind, parallel-group, randomised, active-controlled, phase 3b trial. Lancet 2020, 395, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Helliwell, P.S.; Hjuler, K.F.; Raymond, K.; McInnes, I. Brodalumab in psoriatic arthritis: Results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann. Rheum. Dis. 2021, 80, 185. [Google Scholar] [CrossRef]
- Mitra, A.; Raychaudhuri, S.; Raychaudhuri, S.P. IL-17 and IL-17R: An auspicious therapeutic target for psoriatic disease. Actas Dermo Sifiliogr. 2014, 105, 21–33. [Google Scholar] [CrossRef]
- Kolbinger, F.; Loesche, C.; Valentin, M.-A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F. β-Defensin 2 is a responsive biomarker of IL-17A–driven skin pathology in patients with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923–932.e928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatt, S.; Baeten, D.; Baker, T.; Griffiths, M.; Ionescu, L.; Lawson, A.D.; Maroof, A.; Oliver, R.; Popa, S.; Strimenopoulou, F. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: Evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann. Rheum. Dis. 2018, 77, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.T.; Kavanaugh, A.; Merola, J.F.; Schett, G.; Scher, J.U.; Warren, R.B.; Gottlieb, A.B.; Assudani, D.; Bedford-Rice, K.; Coarse, J. Bimekizumab in patients with active psoriatic arthritis: Results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet 2020, 395, 427–440. [Google Scholar] [CrossRef]
- McInnes, I.B.; Asahina, A.; Coates, L.C.; Landewé, R.; Merola, J.F.; Ritchlin, C.T.; Tanaka, Y.; Gossec, L.; Gottlieb, A.B.; Warren, R.B.; et al. Bimekizumab in patients with psoriatic arthritis, naive to biologic treatment: A randomised, double-blind, placebo-controlled, phase 3 trial (BE OPTIMAL). Lancet 2022, 401, 25–37. [Google Scholar] [CrossRef]
- Merola, J.F.; Landewé, R.; McInnes, I.B.; Mease, P.J.; Ritchlin, C.T.; Tanaka, Y.; Asahina, A.; Behrens, F.; Gladman, D.D.; Gossec, L.; et al. Bimekizumab in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor-α inhibitors: A randomised, double-blind, placebo-controlled, phase 3 trial (BE COMPLETE). Lancet 2023, 401, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Warren, R.B.; Lebwohl, M.; Gooderham, M.; Strober, B.; Langley, R.G.; Paul, C.; De Cuyper, D.; Vanvoorden, V.; Madden, C.; et al. Bimekizumab versus Secukinumab in Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 142–152. [Google Scholar] [CrossRef]
- Warren, R.B.; Blauvelt, A.; Bagel, J.; Papp, K.A.; Yamauchi, P.; Armstrong, A.; Langley, R.G.; Vanvoorden, V.; De Cuyper, D.; Cioffi, C.; et al. Bimekizumab versus Adalimumab in Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 130–141. [Google Scholar] [CrossRef]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Langley, R.G.; Armstrong, A.; Warren, R.B.; Gordon, K.B.; Merola, J.F.; Okubo, Y.; Madden, C.; et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): Efficacy and safety from a 52-week, multicentre, double-blind, active comparator and placebo controlled phase 3 trial. Lancet 2021, 397, 487–498. [Google Scholar] [CrossRef]
- Veale, D.J.; McGonagle, D.; McInnes, I.B.; Krueger, J.G.; Ritchlin, C.T.; Elewaut, D.; Kanik, K.S.; Hendrikx, T.; Berstein, G.; Hodge, J.; et al. The rationale for Janus kinase inhibitors for the treatment of spondyloarthritis. Rheumatology 2019, 58, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E., 3rd; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [Green Version]
- Fragoulis, G.E.; Siebert, S.; McInnes, I.B. Therapeutic Targeting of IL-17 and IL-23 Cytokines in Immune-Mediated Diseases. Annu. Rev. Med. 2016, 67, 337–353. [Google Scholar] [CrossRef]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.; Rigby, W.; Azevedo, V.F.; Behrens, F.; Blanco, R.; Kaszuba, A.; Kudlacz, E.; Wang, C.; Menon, S.; Hendrikx, T. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N. Engl. J. Med. 2017, 377, 1525–1536. [Google Scholar] [CrossRef]
- Liu, K.D.; Gaffen, S.L.; Goldsmith, M.A. JAK/STAT signaling by cytokine receptors. Curr. Opin. Immunol. 1998, 10, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.; Coates, L.C.; Fleishaker, D.; Kivitz, A.J.; Mease, P.J.; Gladman, D.D.; FitzGerald, O.; Wang, C.; Wu, J.; Hsu, M.-A.; et al. Safety and efficacy of tofacitinib up to 48 months in patients with active psoriatic arthritis: Final analysis of the OPAL Balance long-term extension study. Lancet Rheumatol. 2021, 3, e270–e283. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef]
- McInnes, I.B.; Anderson, J.K.; Magrey, M.; Merola, J.F.; Liu, Y.; Kishimoto, M.; Jeka, S.; Pacheco-Tena, C.; Wang, X.; Chen, L.; et al. Trial of Upadacitinib and Adalimumab for Psoriatic Arthritis. N. Engl. J. Med. 2021, 384, 1227–1239. [Google Scholar] [CrossRef]
- Mease, P.J.; Lertratanakul, A.; Anderson, J.K.; Papp, K.; Van den Bosch, F.; Tsuji, S.; Dokoupilova, E.; Keiserman, M.; Wang, X.; Zhong, S.; et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 2021, 80, 312. [Google Scholar] [CrossRef]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef]
- Nogueira, M.; Puig, L.; Torres, T. JAK inhibitors for treatment of psoriasis: Focus on selective TYK2 inhibitors. Drugs 2020, 80, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef] [PubMed]
- A Study to Determine the Efficacy and Safety of Deucravacitinib Compared with Placebo in Participants with Active Psoriatic Arthritis (PsA) Who Are Naïve to Biologic Disease-Modifying Anti-Rheumatic Drugs. Available online: https://ClinicalTrials.gov/show/NCT04908202 (accessed on 19 December 2022).
- A Study to Determine the Efficacy and Safety of Deucravacitinib Compared with Placebo in Participants with Active Psoriatic Arthritis (PsA) Who Are Naïve to Biologic Disease Modifying Anti-Rheumatic Drugs or Had Previously Received TNFα Inhibitor Treatment. Available online: https://ClinicalTrials.gov/show/NCT04908189 (accessed on 19 December 2022).
- Mease, P.J.; Deodhar, A.A.; van der Heijde, D.; Behrens, F.; Kivitz, A.J.; Neal, J.; Kim, J.; Singhal, S.; Nowak, M.; Banerjee, S. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann. Rheum. Dis. 2022, 81, 815. [Google Scholar] [CrossRef]
- Mease, P.J.; Gottlieb, A.B.; van der Heijde, D.; FitzGerald, O.; Johnsen, A.; Nys, M.; Banerjee, S.; Gladman, D.D. Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann. Rheum. Dis. 2017, 76, 1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mease, P.J.; Gottlieb, A.B.; Berman, A.; Drescher, E.; Xing, J.; Wong, R.; Banerjee, S. The Efficacy and Safety of Clazakizumab, an Anti–Interleukin-6 Monoclonal Antibody, in a Phase IIb Study of Adults With Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, A.; Rosenø, N.A.L.; Aagaard, D.; Lørup, E.H.; Nielsen, M.-L.; Nymand, L.; Kristensen, L.E.; Thyssen, J.P.; Thomsen, S.F.; Cordtz, R.L.; et al. Drug survival of biologics and novel immunomodulators for rheumatoid arthritis, axial spondyloarthritis, psoriatic arthritis, and psoriasis—A nationwide cohort study from the DANBIO and DERMBIO registries. Semin. Arthritis Rheum. 2022, 53, 151979. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Turk, M.; Alammari, Y.; Young, F.; Gallagher, P.; Saber, T.; Fearon, U.; Veale, D.J. Long-term remission and biologic persistence rates: 12-year real-world data. Arthritis Res. Ther. 2021, 23, 25. [Google Scholar] [CrossRef]
- Lindström, U.; Di Giuseppe, D.; Delcoigne, B.; Glintborg, B.; Möller, B.; Ciurea, A.; Pombo-Suarez, M.; Sanchez-Piedra, C.; Eklund, K.; Relas, H.; et al. Effectiveness and treatment retention of TNF inhibitors when used as monotherapy versus comedication with csDMARDs in 15 332 patients with psoriatic arthritis. Data from the EuroSpA collaboration. Ann. Rheum. Dis. 2021, 80, 1410. [Google Scholar] [CrossRef]
- Pina Vegas, L.; Penso, L.; Claudepierre, P.; Sbidian, E. Long-term Persistence of First-line Biologics for Patients With Psoriasis and Psoriatic Arthritis in the French Health Insurance Database. JAMA Dermatol. 2022, 158, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, I.; Nakayamada, S.; Nakano, K.; Kubo, S.; Iwata, S.; Miyazaki, Y.; Yoshikawa, M.; Yoshinari, H.; Tanaka, Y. Precision medicine using different biological DMARDs based on characteristic phenotypes of peripheral T helper cells in psoriatic arthritis. Rheumatology 2019, 58, 336–344. [Google Scholar] [CrossRef]
- Kristensen, L.E.; Keiserman, M.; Papp, K.; McCasland, L.; White, D.; Lu, W.; Wang, Z.; Soliman, A.M.; Eldred, A.; Barcomb, L.; et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann. Rheum. Dis. 2022, 81, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Östör, A.; Van den Bosch, F.; Papp, K.; Asnal, C.; Blanco, R.; Aelion, J.; Alperovich, G.; Lu, W.; Wang, Z.; Soliman, A.M.; et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann. Rheum. Dis. 2022, 81, 351. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Therapy | Mechanism of Action |
|---|---|
| Infliximab | Chimeric monoclonal antibody against TNF-α |
| Etanercept | Soluble TNF receptor p75-immunoglobulin G1 fusion protein |
| Adalimumab | Fully human anti-TNF-α monoclonal antibody |
| Golimumab | Fully human immunoglobulin G1 kappa anti-TNF-α antibody |
| Certolizumab pegol | Fab fragment of anti-TNF- α monoclonal antibody |
| Ustekinumab | Fully human immunoglobulin G1 kappa monoclonal antibody against the shared p40 subunit of IL-12 and IL-23 |
| Guselkumab | Fully human immunoglobulin G1 lambda monoclonal antibody against the p19 subunit of IL-23 |
| Risankizumab | Fully human immunoglobulin G1 lambda monoclonal antibody against the p19 subunit of IL-23 |
| Secukinumab | Fully human immunoglobulin G1 kappa monoclonal antibody against IL-17A |
| Ixekizumab | Humanised immunoglobulin G4 monoclonal antibody against IL-17A |
| Brodalumab | Fully human immunoglobulin G2 monoclonal antibody against IL-17RA |
| Bimekizumab | Humanised immunoglobulin G1 monoclonal antibody against IL-17A and IL-17F |
| Tofacitinib | Inhibitor of JAK1 and JAK3 |
| Upadacitinib | Selective inhibitor of JAK1 |
| Filgotinib | Selective inhibitor of JAK1 |
| Deucravacitinib | Selective TYK2 inhibitor |
| Abatacept | Selective T cell co-stimulator inhibitor |
| Study | Comparison | Population | TNFi-IR (%) | MTX Use (%) | Primary Outcome | Primary Endpoint (Weeks) | ACR20 (%) |
|---|---|---|---|---|---|---|---|
| TNF-α inhibitors | |||||||
| Antoni et al. (IMPACT 2) (N = 200) [36] | IFX vs. PBO | csDMARD-IR or NSAID-IR | 0 | 46 | ACR20 | 14 | 58 (IFX) vs. 11 (PBO) * |
| Mease et al. (N = 205) [35] | ETN vs. PBO | NSAID-IR | 0 | 41 | ACR20 | 12 | 59 (ETN) vs. 15 (PBO) * |
| Mease et al. (ADEPT) (N = 313) [34] | ADA vs. PBO | NSAID-IR | 0 | 50.5 | ACR20 | 12 | 58(ADA) vs. 14 (PBO) * |
| Kavanaugh et al. (GO-REVEAL) (N = 405) [37] | GOL (50 mg, 100 mg) vs. PBO | NSAID-IR | 0 | 48 | ACR20 | 24 | 48 (combined) vs. 9 (PBO) * |
| Mease et al. (RAPID-PsA) (N = 409) [38] | CTZ (200 mg Q2W, 400 mg Q4W) vs. PBO | csDMARD-IR | NR 38.9 prior TNFi | 64 | ACR20 | 12 | 58 (Q2W) 51.9 (Q4W) vs. 24.3 (PBO) * |
| IL-12, 23p40 inhibitor | |||||||
| McInnes et al. (PSUMMIT 1) (N = 615) [55] | UST (45 mg, 90 mg) vs. PBO | csDMARD-IR | 0 | 48 | ACR20 | 24 | 42.4 (45 mg) 49.5 (90 mg) 22.8 (PBO) * |
| Ritchlin et al. (PSUMMIT 2) (N = 312) [56] | UST (45 mg, 90 mg) vs. PBO | Mixed csDMARD-IR/TNFi-IR | 57.7 | 50 | ACR20 | 24 | 43.8 (combined) 20.2 (PBO) * |
| Il-23p19 inhibitors | |||||||
| Deodhar et al. (DISCOVER-1) (N = 381) [67] | GKM (Q4W, Q8W) vs. PBO | Mixed csDMARD-IR/Apremilast-IR/TNFi-IR | 12 | 55 | ACR20 | 24 | 59 (Q4W) 52 (Q8W) 22 (PBO) * |
| Mease et al. (DISCOVER-2) (N = 739) [68] | GKM (Q4W, Q8W) vs. PBO | csDMARD-IR | 0 | 60 | ACR20 | 24 | 64 (Q4W) 64 (Q8W) 33 (PBO) * |
| Coates et al. (COSMOS) (N = 285) [73] | GKM vs. PBO | TNFi-IR | 84 | 55 | ACR20 | 24 | 44.4 (GKM) vs. 19.8 (PBO) * |
| Kristensen et al. (KEEPsAKE-1) (N = 964) [141] | RKM vs. PBO | csDMARD-IR | 0 | 65.2 | ACR20 | 24 | 57.3 (RKM) vs. 33.5 (PBO) * |
| Östör et al. (KEEPsAKE-2) (N = 443) [142] | RKM vs. PBO | Mixed csDMARD-IR/Bio-IR | 46 | 47.1 | ACR20 | 24 | 51.3 (RKM) vs. 26.5 (PBO) * |
| Il-17A inhibitors | |||||||
| Mease et al. (FUTURE 1) (N = 606) [88] | SEC (150 mg, 75 mg) vs. PBO | Mixed csDMARD-IR/TNFi-IR | 29.4 | 61 | ACR20 | 24 | 50 (150 mg) 50.5 (75 mg) 17.3 (PBO) |
| McInnes et al. (FUTURE 2) (N = 397) [89] | SEC (300 mg, 150 mg, 75 mg) vs. PBO | Mixed csDMARD-IR/TNFi-IR | 35 | 47 | ACR20 | 24 | 54 (300 mg) 51 (150 mg) 29 (75 mg) 15 (PBO) * |
| McInnes et al. (EXCEED) (N = 853) [107] | SEC vs. ADA | csDMARD-IR | 0 | 85 | ACR20 | 52 | 67 (SEC) 62 (ADA) |
| Mease et al. (SPIRIT-P1) (N = 417) [103] | IXE (Q2W, Q4W) vs. PBO vs. ADA (reference arm) | csDMARD-IR | 0 | 54.2 | ACR20 | 24 | 62.1 (Q2W) 57.9 (Q4W) 30.2 (PBO) * 57.4 (ADA) |
| Nash et al. (SPIRIT-P2) (N = 363) [102] | IXE (Q2W, Q4W) vs. PBO | TNFi-IR | 91 | 41 | ACR20 | 24 | 48 (Q2W) 53 (Q4W) 20 (PBO) * |
| IL-17RA inhibitor | |||||||
| Mease et al. (AMVISION-1 and AMVISION-2) (N = 962) [108] | Brodalumab (140 mg, 210 mg) vs. PBO | Mixed csDMARD-IR/Bio-IR | NR | NR | ACR20 | 16 | 45.8 (140 mg) 47.9 (210 mg) 20.9 (PBO) * |
| IL-17A/F inhibitor | |||||||
| McInnes et al. (BE-OPTIMAL) (N = 852) [113] | BKM vs. PBO vs. ADA (reference arm) | csDMARD-IR | 0 | 58 | ACR50 (44% BKZ vs. 10% PBO) * | 16 | 62.2 (BKZ) vs. 24 (PBO) * 68.6 (ADA) |
| Merola et al. (BE-COMPLETE) (N = 400) [114] | BKM vs. PBO | TNFi-IR | 88 | 43 | ACR50 (43% BKZ vs. 7% PBO) * | 16 | 67 (BKZ) vs. 15.8 (PBO) * |
| JAK inhibitors | |||||||
| Mease et al. (OPAL Broaden) (N = 422) [121] | TOFA (5 mg BD, 10 mg BD) vs. PBO vs. ADA (reference arm) | csDMARD-IR | 0 | 84 | ACR20 | 12 | 50 (5 mg) vs. 61 (10 mg) vs. 33 (PBO) * 52% (ADA) |
| Gladman et al. (OPAL Beyond) (N = 394) [122] | TOFA (5 mg BD, 10 mg BD) vs. PBO | TNFi-IR | 100 | 73 | ACR20 | 12 | 50 (5 mg) vs. 47 (10 mg) vs. 24 (PBO) * |
| McInnes et al. (SELECT-PsA 1) (N = 1704) [126] | UPA (15 mg, 30 mg) vs. PBO vs. ADA (reference arm) | csDMARD-IR | 0 | 84 | ACR20 | 12 | 70.6 (15 mg) vs. 78.5 (30 mg) vs. 36.2 (PBO) * 65 (ADA) |
| Mease et al. (SELECT-PSA 2) (N = 641) [127] | UPA (15 mg, 30 mg) vs. PBO | Bio-IR | NR 92% Bio-IR | 37 | ACR20 | 12 | 56.9 (15 mg) vs. 63.8 (30 mg) vs. 24.1 (PBO) * |
| Mease et al. (EQUATOR) (N = 131) [128] | Filgotinib vs. PBO | csDMARD-IR | 0 | 55 | ACR20 | 16 | 80 (filgotinib) vs. 33 (PBO) * |
| Other targets | |||||||
| Mease et al. (N = 203) [133] | Deucravacitinib (6 mg, 12 mg) vs. PBO | Mixed csDMARD-IR/TNFi-IR | NR 15.8% prior TNFi | 54.7 | ACR20 | 16 | 52.9 (6 mg) vs. 62.7 (12 mg) vs. 31.8 (PBO) * |
| Mease et al. (ASTRAEA) (N = 424) [134] | ABA vs. PBO | Mixed csDMARD-IR/TNFi-IR | 61 | 60 | ACR20 | 24 | 39.4 (ABA) vs. 22.3 (PBO) * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundanum, S.; Orr, C.; Veale, D. Targeted Therapies in Psoriatic Arthritis—An Update. Int. J. Mol. Sci. 2023, 24, 6384. https://doi.org/10.3390/ijms24076384
Sundanum S, Orr C, Veale D. Targeted Therapies in Psoriatic Arthritis—An Update. International Journal of Molecular Sciences. 2023; 24(7):6384. https://doi.org/10.3390/ijms24076384
Chicago/Turabian StyleSundanum, Sonia, Carl Orr, and Douglas Veale. 2023. "Targeted Therapies in Psoriatic Arthritis—An Update" International Journal of Molecular Sciences 24, no. 7: 6384. https://doi.org/10.3390/ijms24076384