
The Role of LFA-1 for the Differentiation and Function of Regulatory T Cells—Lessons Learned from Different Transgenic Mouse Models

{kind=link}
{kind=link}
Abstract
:1. Treg Subsets and Functions within the Immune System
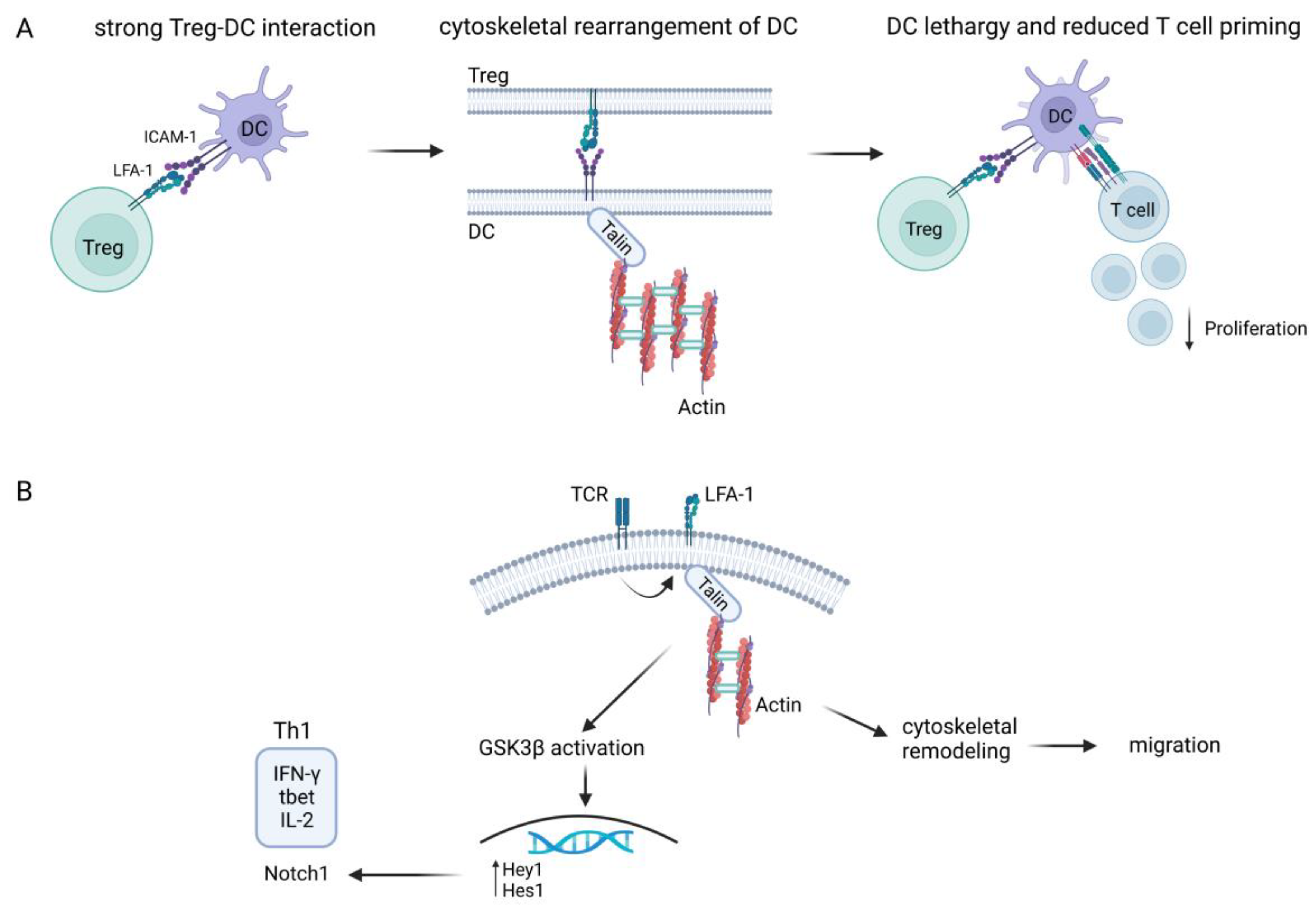
2. Role of LFA-1 on T Cells
3. Leukocyte Adhesion Deficiency Type-1 (LAD-1)
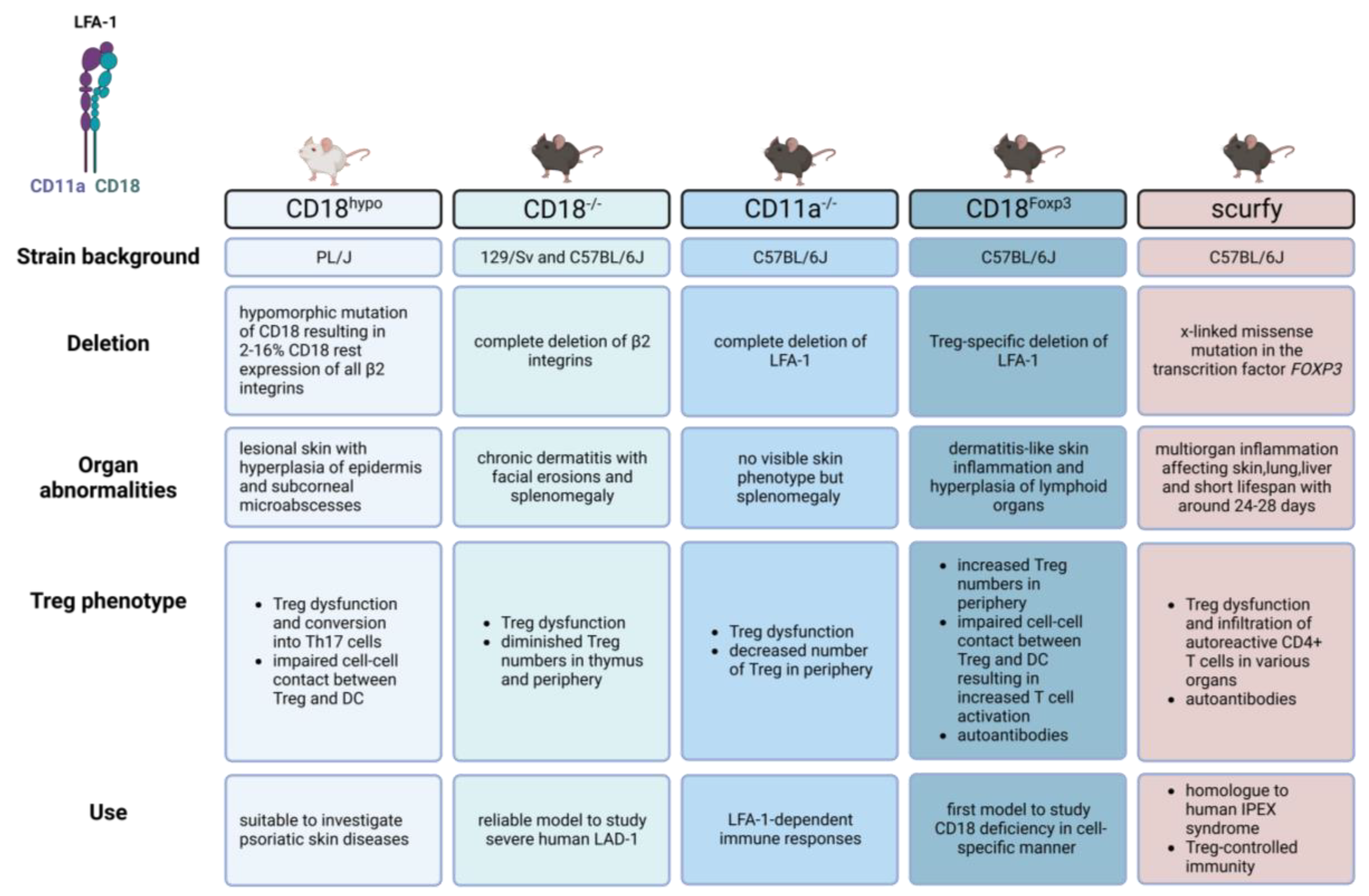
4. β2-integrin-Deficient Mouse Models
4.1. CD18 Hypomorphic Mouse (CD18hypo)
4.2. CD18 Null Mouse (CD18−/−)
4.3. LFA-1 Null Mouse (CD11a−/−)
4.4. CD18Foxp3 Mouse
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2019, 10, 3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocamora-Reverte, L.; Melzer, F.L.; Wurzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2020, 11, 616949. [Google Scholar] [CrossRef]
- Mahmud, S.A.; Manlove, L.S.; Schmitz, H.M.; Xing, Y.; Wang, Y.; Owen, D.L.; Schenkel, J.M.; Boomer, J.S.; Green, J.M.; Yagita, H.; et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 2014, 15, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Dejaco, C.; Duftner, C.; Grubeck-Loebenstein, B.; Schirmer, M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006, 117, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Gerard, A.; Cope, A.P.; Kemper, C.; Alon, R.; Kochl, R. LFA-1 in T cell priming, differentiation, and effector functions. Trends Immunol. 2021, 42, 706–722. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef]
- Barzaghi, F.; Passerini, L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front. Pediatr. 2021, 9, 612760. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Bevan, M.J. The nature of the peptide/MHC ligand involved in positive selection. Semin. Immunol. 1996, 8, 63–68. [Google Scholar] [CrossRef]
- Yadav, M.; Stephan, S.; Bluestone, J.A. Peripherally induced tregs—Role in immune homeostasis and autoimmunity. Front. Immunol. 2013, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Okamura, T.; Yamamoto, K.; Fujio, K. Early Growth Response Gene 2-Expressing CD4+LAG3+ Regulatory T Cells: The Therapeutic Potential for Treating Autoimmune Diseases. Front. Immunol. 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.M.S.; Gomes, S.T.M.; Ishak, R.; Vallinoto, A.C.R. Regulatory T Cell and Forkhead Box Protein 3 as Modulators of Immune Homeostasis. Front. Immunol. 2017, 8, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, T.; Fujio, K.; Shibuya, M.; Sumitomo, S.; Shoda, H.; Sakaguchi, S.; Yamamoto, K. CD4+CD25−LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl. Acad. Sci. USA 2009, 106, 13974–13979. [Google Scholar] [CrossRef] [Green Version]
- Bacchetta, R.; Bigler, M.; Touraine, J.L.; Parkman, R.; Tovo, P.A.; Abrams, J.; de Waal Malefyt, R.; de Vries, J.E.; Roncarolo, M.G. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J. Exp. Med. 1994, 179, 493–502. [Google Scholar] [CrossRef]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Pot, C.; Apetoh, L.; Kuchroo, V.K. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin. Immunol. 2011, 23, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Barrat, F.J.; Cua, D.J.; Boonstra, A.; Richards, D.F.; Crain, C.; Savelkoul, H.F.; de Waal-Malefyt, R.; Coffman, R.L.; Hawrylowicz, C.M.; O’Garra, A. In vitro generation of interleukin 10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002, 195, 603–616. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Draghici, E.; Gregori, S.; Mocchetti, C.; Bonifacio, E.; Roncarolo, M.G. Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes 2006, 55, 40–49. [Google Scholar] [CrossRef]
- Delacher, M.; Imbusch, C.D.; Weichenhan, D.; Breiling, A.; Hotz-Wagenblatt, A.; Trager, U.; Hofer, A.C.; Kagebein, D.; Wang, Q.; Frauhammer, F.; et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017, 18, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Delacher, M.; Imbusch, C.D.; Hotz-Wagenblatt, A.; Mallm, J.P.; Bauer, K.; Simon, M.; Riegel, D.; Rendeiro, A.F.; Bittner, S.; Sanderink, L.; et al. Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF. Immunity 2020, 52, 295–312.e211. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Min, B. Tissue Resident Foxp3+ Regulatory T Cells: Sentinels and Saboteurs in Health and Disease. Front. Immunol. 2022, 13, 865593. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Cao, D.; van Vollenhoven, R.; Klareskog, L.; Trollmo, C.; Malmstrom, V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res. Ther. 2004, 6, R335–R346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.F.; Wang, C.R.; Fung, L.L.; Wu, C.R. Decreased CD4+CD25+ T cells in peripheral blood of patients with systemic lupus erythematosus. Scand. J. Immunol. 2004, 59, 198–202. [Google Scholar] [CrossRef]
- Crispin, J.C.; Martinez, A.; Alcocer-Varela, J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J. Autoimmun. 2003, 21, 273–276. [Google Scholar] [CrossRef]
- Furuno, K.; Yuge, T.; Kusuhara, K.; Takada, H.; Nishio, H.; Khajoee, V.; Ohno, T.; Hara, T. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J. Pediatr. 2004, 145, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Powrie, F.; Carlino, J.; Leach, M.W.; Mauze, S.; Coffman, R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 1996, 183, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Gondek, D.C.; Lu, L.F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Tollefsen, B.L.; Kemper, C.; Atkinson, J.P.; Ley, T.J. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004, 104, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [Green Version]
- Safinia, N.; Scotta, C.; Vaikunthanathan, T.; Lechler, R.I.; Lombardi, G. Regulatory T Cells: Serious Contenders in the Promise for Immunological Tolerance in Transplantation. Front. Immunol. 2015, 6, 438. [Google Scholar] [CrossRef] [Green Version]
- Read, S.; Malmstrom, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Duhen, T.; Duhen, R.; Lanzavecchia, A.; Sallusto, F.; Campbell, D.J. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 2012, 119, 4430–4440. [Google Scholar] [CrossRef] [Green Version]
- Wohler, J.; Bullard, D.; Schoeb, T.; Barnum, S. LFA-1 is critical for regulatory T cell homeostasis and function. Mol. Immunol. 2009, 46, 2424–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. beta2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marski, M.; Kandula, S.; Turner, J.R.; Abraham, C. CD18 is required for optimal development and function of CD4+CD25+ T regulatory cells. J. Immunol. 2005, 175, 7889–7897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, L.; Walling, M.K. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol. 2011, 11, 416–426. [Google Scholar] [CrossRef]
- Shimaoka, M.; Takagi, J.; Springer, T.A. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 485–516. [Google Scholar] [CrossRef] [Green Version]
- Verma, N.K.; Kelleher, D. Not Just an Adhesion Molecule: LFA-1 Contact Tunes the T Lymphocyte Program. J. Immunol. 2017, 199, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ganguly, A.; Mucsi, A.D.; Meng, J.; Yan, J.; Detampel, P.; Munro, F.; Zhang, Z.; Wu, M.; Hari, A.; et al. Strong adhesion by regulatory T cells induces dendritic cell cytoskeletal polarization and contact-dependent lethargy. J. Exp. Med. 2017, 214, 327–338. [Google Scholar] [CrossRef]
- Verhagen, J.; Wraith, D.C. Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3+ Treg cells. J. Immunol. Methods 2014, 414, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, N.K.; Dempsey, E.; Long, A.; Davies, A.; Barry, S.P.; Fallon, P.G.; Volkov, Y.; Kelleher, D. Leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction induces a novel genetic signature resulting in T-cells refractory to transforming growth factor-beta signaling. J. Biol. Chem. 2012, 287, 27204–27216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, N.K.; Fazil, M.H.; Ong, S.T.; Chalasani, M.L.; Low, J.H.; Kottaiswamy, A.; Praseetha, P.; Kizhakeyil, A.; Kumar, S.; Panda, A.K.; et al. LFA-1/ICAM-1 Ligation in Human T Cells Promotes Th1 Polarization through a GSK3beta Signaling-Dependent Notch Pathway. J. Immunol. 2016, 197, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K. The second touch hypothesis: T cell activation, homing and polarization. F1000Res 2014, 3, 37. [Google Scholar] [CrossRef]
- Cox, D.P.; Weathers, D.R. Leukocyte adhesion deficiency type 1: An important consideration in the clinical differential diagnosis of prepubertal periodontitis. A case report and review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2008, 105, 86–90. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Van Lier, R.A.; Hamann, D.; de Boer, M.; Thung, L.Y.; Weening, R.S.; Verhoeven, A.J.; Roos, D. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J. Clin. Investig. 1997, 100, 1725–1733. [Google Scholar] [CrossRef] [Green Version]
- Kambli, P.M.; Bargir, U.A.; Yadav, R.M.; Gupta, M.R.; Dalvi, A.D.; Hule, G.; Kelkar, M.; Sawant-Desai, S.; Setia, P.; Jodhawat, N.; et al. Clinical and Genetic Spectrum of a Large Cohort of Patients With Leukocyte Adhesion Deficiency Type 1 and 3: A Multicentric Study From India. Front. Immunol. 2020, 11, 612703. [Google Scholar] [CrossRef]
- Almarza Novoa, E.; Kasbekar, S.; Thrasher, A.J.; Kohn, D.B.; Sevilla, J.; Nguyen, T.; Schwartz, J.D.; Bueren, J.A. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 2018, 6, 1418–1420.e1410. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; van Bruggen, R.; Kamerbeek, N.; Tool, A.T.; Hicsonmez, G.; Gurgey, A.; Karow, A.; Verhoeven, A.J.; Seeger, K.; Sanal, O.; et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood 2007, 109, 3529–3537. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, D.; Regairaz, L.; Deswarte, C.; Garcia, M.; Richard, M.E.; Casanova, J.L.; Bustamante, J.; Perez, L. Leukocyte Adhesion Deficiency Type 1 (LAD1) with Expressed but Nonfunctional CD11/CD18. J. Clin. Immunol. 2016, 36, 627–630. [Google Scholar] [CrossRef]
- De Rose, D.U.; Giliani, S.; Notarangelo, L.D.; Lougaris, V.; Lanfranchi, A.; Moratto, D.; Martire, B.; Specchia, F.; Tommasini, A.; Plebani, A.; et al. Long term outcome of eight patients with type 1 Leukocyte Adhesion Deficiency (LAD-1): Not only infections, but high risk of autoimmune complications. Clin. Immunol. 2018, 191, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Uzel, G.; Kleiner, D.E.; Kuhns, D.B.; Holland, S.M. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology 2001, 121, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Justiz, V. Leukocyte Adhesion Deficiency; SatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Uzel, G.; Tng, E.; Rosenzweig, S.D.; Hsu, A.P.; Shaw, J.M.; Horwitz, M.E.; Linton, G.F.; Anderson, S.M.; Kirby, M.R.; Oliveira, J.B.; et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1). Blood 2008, 111, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W.; Cavazzana-Calvo, M.; Davies, E.G.; Davis, J.; Duval, M.; Eames, G.; Farinha, N.; Filopovich, A.; Fischer, A.; Friedrich, W.; et al. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics 2009, 123, 836–840. [Google Scholar] [CrossRef] [Green Version]
- Tran, D. Dysfunctional FOXP3 + Regulatory T cells in Leukocyte Adhesion Deficiency Type 1 (LAD-1) Patients with Reversion Mutations and Inflammatory Bowel Disease. J. Allergy Clin. Immunol. 2009, 123, S15. [Google Scholar] [CrossRef]
- Rosa, J.P.; McEver, R.P. Processing and assembly of the integrin, glycoprotein IIb-IIIa, in HEL cells. J. Biol. Chem. 1989, 264, 12596–12603. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.W.; Ballantyne, C.M.; Smith, C.W.; Montgomery, C.; Bradley, A.; O’Brien, W.E.; Beaudet, A.L. Gene targeting yields a CD18-mutant mouse for study of inflammation. J. Immunol. 1993, 151, 1571–1578. [Google Scholar] [CrossRef]
- Bullard, D.C.; Scharffetter-Kochanek, K.; McArthur, M.J.; Chosay, J.G.; McBride, M.E.; Montgomery, C.A.; Beaudet, A.L. A polygenic mouse model of psoriasiform skin disease in CD18-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2116–2121. [Google Scholar] [CrossRef] [Green Version]
- Peters, T.; Sindrilaru, A.; Wang, H.; Oreshkova, T.; Renkl, A.C.; Kess, D.; Scharffetter-Kochanek, K. CD18 in monogenic and polygenic inflammatory processes of the skin. J. Investig. Dermatol. Symp. Proc. 2006, 11, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced CD18 levels drive regulatory T cell conversion into Th17 cells in the CD18hypo PL/J mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Peters, T.; Sindrilaru, A.; Kess, D.; Oreshkova, T.; Yu, X.Z.; Seier, A.M.; Schreiber, H.; Wlaschek, M.; Blakytny, R.; et al. TGF-beta-dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J. Clin. Investig. 2008, 118, 2629–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, N.; Sodani, R.; Chandra, J.; Singh, V. Leukocyte adhesion defect type 1 presenting with recurrent pyoderma gangrenosum. Indian J. Dermatol. 2013, 58, 158. [Google Scholar] [CrossRef]
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204. [Google Scholar] [CrossRef]
- El-Sayed, Z.A.; El-Ghoneimy, D.H.; Abd-Allah, H.; Afifi, H.M. A rare association between leukocyte adhesion deficiency type I and psoriasis in humans. Allergy Asthma Immunol. Res. 2011, 3, 138–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Pelt, J.P.; Kuijpers, S.H.; van de Kerkhof, P.C.; de Jong, E.M. The CD11b/CD18-integrin in the pathogenesis of psoriasis. J. Dermatol. Sci. 1998, 16, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Scharffetter-Kochanek, K.; Lu, H.; Norman, K.; van Nood, N.; Munoz, F.; Grabbe, S.; McArthur, M.; Lorenzo, I.; Kaplan, S.; Ley, K.; et al. Spontaneous Skin Ulceration and Defective T Cell Function in CD18 Null Mice. J. Exp. Med. 1998, 188, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Kess, D.; Peters, T.; Zamek, J.; Wickenhauser, C.; Tawadros, S.; Loser, K.; Varga, G.; Grabbe, S.; Nischt, R.; Sunderkotter, C.; et al. CD4+ T cell-associated pathophysiology critically depends on CD18 gene dose effects in a murine model of psoriasis. J. Immunol. 2003, 171, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Grabbe, S.; Varga, G.; Beissert, S.; Steinert, M.; Pendl, G.; Seeliger, S.; Bloch, W.; Peters, T.; Schwarz, T.; Sunderkötter, C.; et al. β2 integrins are required for skin homing of primed T cells but not for priming naive T cells. J. Clin. Investig. 2002, 109, 183–192. [Google Scholar] [CrossRef]
- Peters, T.; Sindrilaru, A.; Hinz, B.; Hinrichs, R.; Menke, A.; Al-Azzeh, E.A.; Holzwarth, K.; Oreshkova, T.; Wang, H.; Kess, D.; et al. Wound-healing defect of CD18−/− mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J. 2005, 24, 3400–3410. [Google Scholar] [CrossRef] [Green Version]
- Schmits, R.; Kundig, T.M.; Baker, D.M.; Shumaker, G.; Simard, J.J.; Duncan, G.; Wakeham, A.; Shahinian, A.; van der Heiden, A.; Bachmann, M.F.; et al. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J. Exp. Med. 1996, 183, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 2008, 105, 10113–10118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gultner, S.; Kuhlmann, T.; Hesse, A.; Weber, J.P.; Riemer, C.; Baier, M.; Hutloff, A. Reduced Treg frequency in LFA-1-deficient mice allows enhanced T effector differentiation and pathology in EAE. Eur. J. Immunol. 2010, 40, 3403–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarczyk, M.; Bolduan, V.; Haist, M.; Stege, H.; Hieber, C.; Johann, L.; Schelmbauer, C.; Blanfeld, M.; Karram, K.; Schunke, J.; et al. beta2 Integrins on Dendritic Cells Modulate Cytokine Signaling and Inflammation-Associated Gene Expression, and Are Required for Induction of Autoimmune Encephalomyelitis. Cells 2022, 11, 2188. [Google Scholar] [CrossRef] [PubMed]
- Klaus, T.; Wilson, A.S.; Vicari, E.; Hadaschik, E.; Klein, M.; Helbich, S.S.C.; Kamenjarin, N.; Hodapp, K.; Schunke, J.; Haist, M.; et al. Impaired Treg-DC interactions contribute to autoimmunity in leukocyte adhesion deficiency type 1. JCI Insight 2022, 7, e162580. [Google Scholar] [CrossRef] [PubMed]
- Andrew, D.P.; Spellberg, J.P.; Takimoto, H.; Schmits, R.; Mak, T.W.; Zukowski, M.M. Transendothelial migration and trafficking of leukocytes in LFA-1-deficient mice. Eur. J. Immunol. 1998, 28, 1959–1969. [Google Scholar] [CrossRef]
- Yilmaz, O.K.; Haeberle, S.; Zhang, M.; Fritzler, M.J.; Enk, A.H.; Hadaschik, E.N. Scurfy Mice Develop Features of Connective Tissue Disease Overlap Syndrome and Mixed Connective Tissue Disease in the Absence of Regulatory T Cells. Front. Immunol. 2019, 10, 881. [Google Scholar] [CrossRef]
- Bennett, C.L.; Brunkow, M.E.; Ramsdell, F.; O’Briant, K.C.; Zhu, Q.; Fuleihan, R.L.; Shigeoka, A.O.; Ochs, H.D.; Chance, P.F. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 2001, 53, 435–439. [Google Scholar] [CrossRef]
- Hoffman, I.E.; Peene, I.; Meheus, L.; Huizinga, T.W.; Cebecauer, L.; Isenberg, D.; De Bosschere, K.; Hulstaert, F.; Veys, E.M.; De Keyser, F. Specific antinuclear antibodies are associated with clinical features in systemic lupus erythematosus. Ann. Rheum. Dis. 2004, 63, 1155–1158. [Google Scholar] [CrossRef]
- Lipsky, D.S.; Lipsky, P.E. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2020, 16, 565–579. [Google Scholar]
- Stochmal, A.; Czuwara, J.; Trojanowska, M.; Rudnicka, L. Antinuclear Antibodies in Systemic Sclerosis: An Update. Clin. Rev. Allergy Immunol. 2020, 58, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Sajda, T.; Sinha, A.A. Autoantibody Signaling in Pemphigus Vulgaris: Development of an Integrated Model. Front. Immunol. 2018, 9, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Di Zenzo, G.; Cozzani, E.; Berti, E.; Cugno, M.; Marzano, A.V. New Insights Into the Pathogenesis of Bullous Pemphigoid: 2019 Update. Front. Immunol. 2019, 10, 1506. [Google Scholar] [CrossRef]
- Varga, G.; Nippe, N.; Balkow, S.; Peters, T.; Wild, M.K.; Seeliger, S.; Beissert, S.; Krummen, M.; Roth, J.; Sunderkotter, C.; et al. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J. Investig. Dermatol. 2010, 130, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Haist, M.; Ries, F.; Gunzer, M.; Bednarczyk, M.; Siegel, E.; Kuske, M.; Grabbe, S.; Radsak, M.; Bros, M.; Teschner, D. Neutrophil-Specific Knockdown of beta2 Integrins Impairs Antifungal Effector Functions and Aggravates the Course of Invasive Pulmonal Aspergillosis. Front. Immunol. 2022, 13, 823121. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaus, T.; Wilson, A.; Fichter, M.; Bros, M.; Bopp, T.; Grabbe, S. The Role of LFA-1 for the Differentiation and Function of Regulatory T Cells—Lessons Learned from Different Transgenic Mouse Models. Int. J. Mol. Sci. 2023, 24, 6331. https://doi.org/10.3390/ijms24076331
Klaus T, Wilson A, Fichter M, Bros M, Bopp T, Grabbe S. The Role of LFA-1 for the Differentiation and Function of Regulatory T Cells—Lessons Learned from Different Transgenic Mouse Models. International Journal of Molecular Sciences. 2023; 24(7):6331. https://doi.org/10.3390/ijms24076331
Chicago/Turabian StyleKlaus, Tanja, Alicia Wilson, Michael Fichter, Matthias Bros, Tobias Bopp, and Stephan Grabbe. 2023. "The Role of LFA-1 for the Differentiation and Function of Regulatory T Cells—Lessons Learned from Different Transgenic Mouse Models" International Journal of Molecular Sciences 24, no. 7: 6331. https://doi.org/10.3390/ijms24076331