
The Novel Compound SUL-138 Counteracts Endothelial Cell and Kidney Dysfunction in Sepsis by Preserving Mitochondrial Function
 , and
, and
Abstract
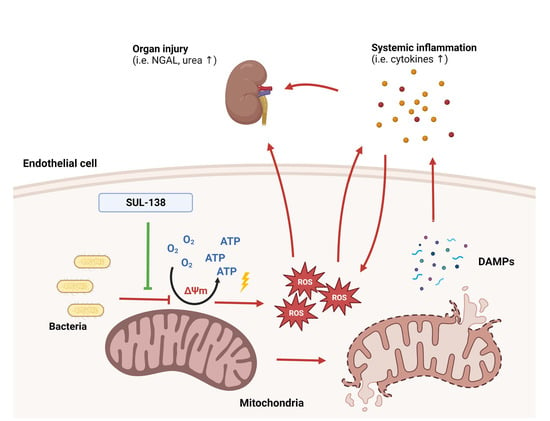
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
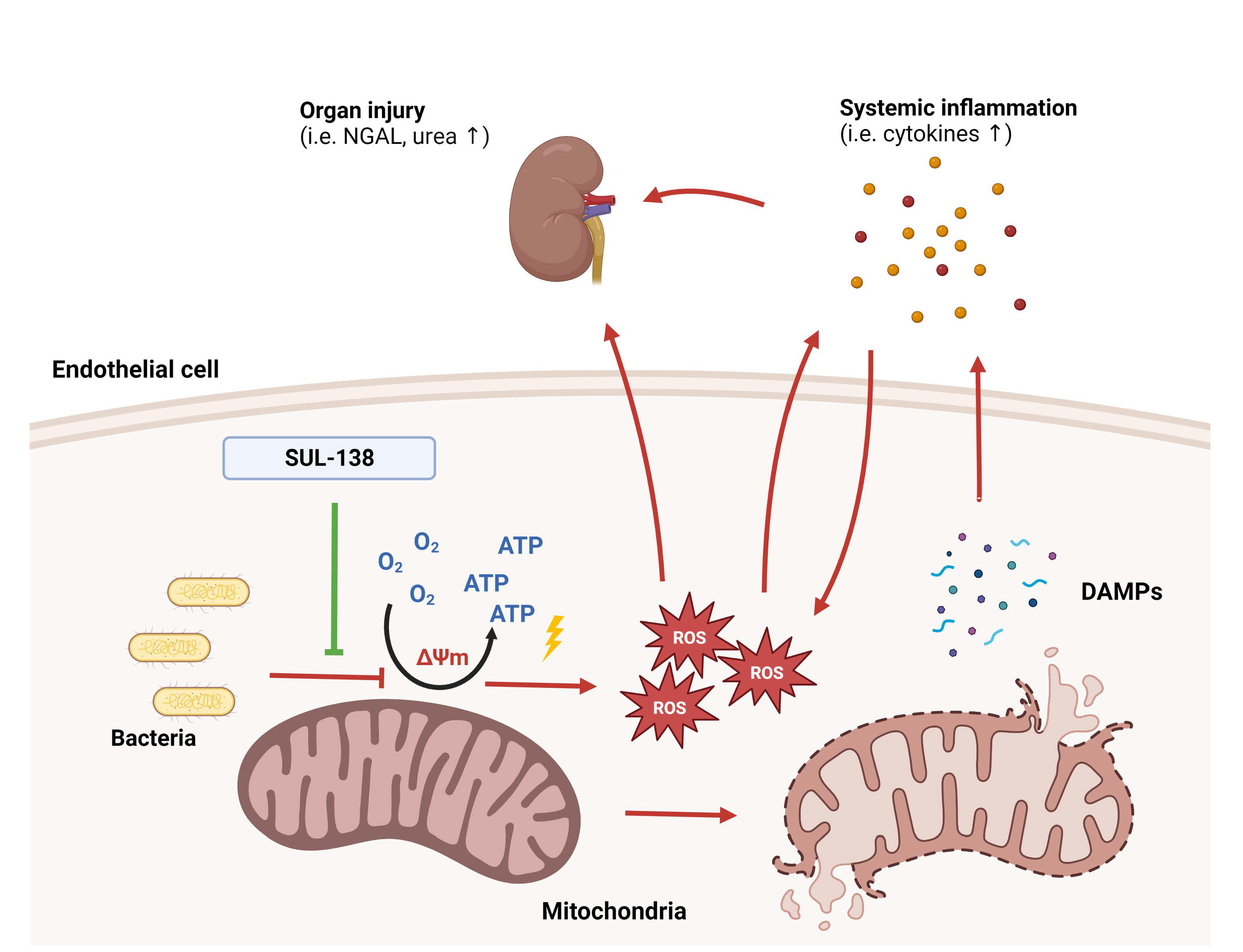
2.1. Effects of SUL-138 on Mitochondria and Survival in LPS-Treated Endothelial Cells
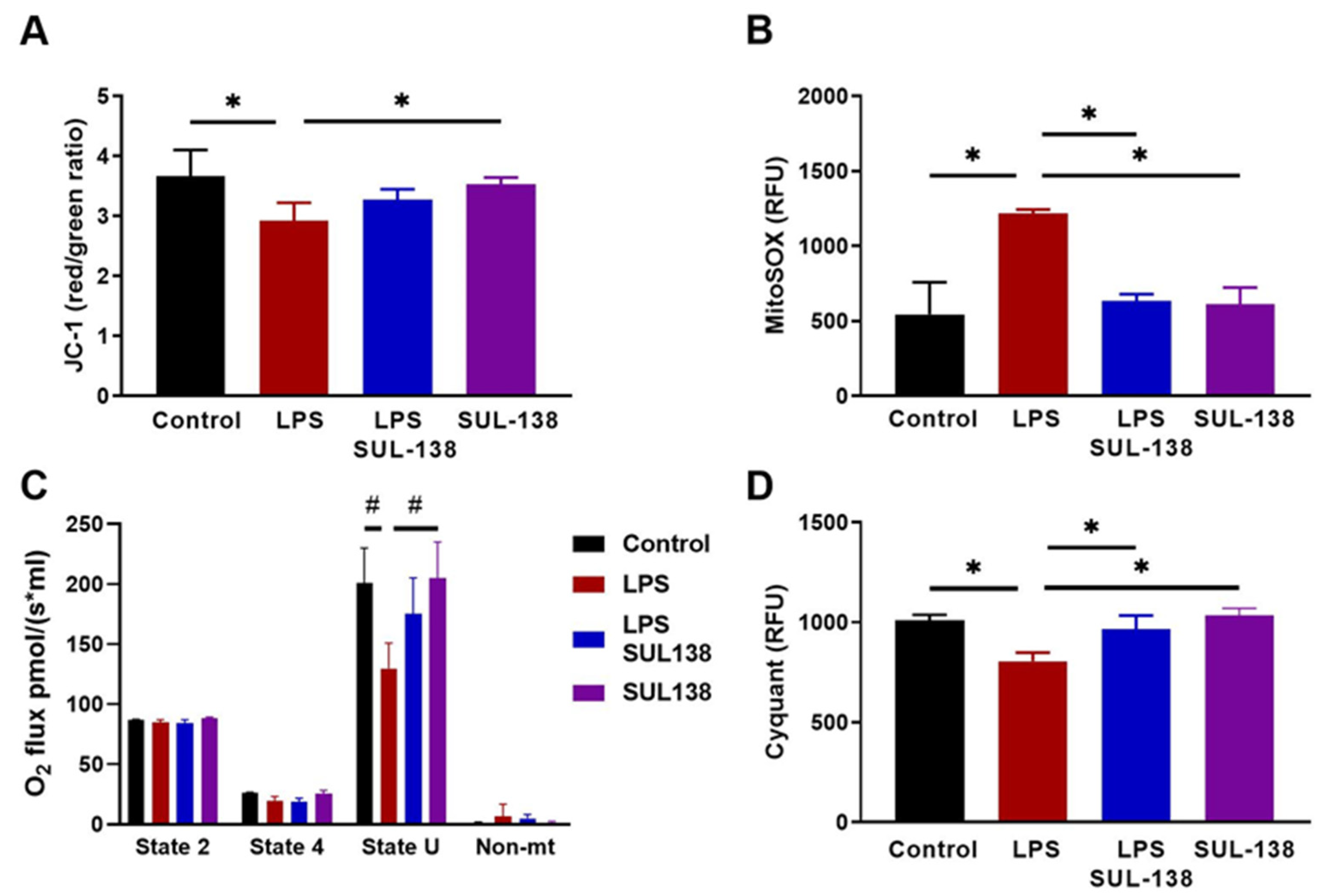
2.2. Effects of SUL-138 on Inflammation in LPS-Treated Endothelial Cells
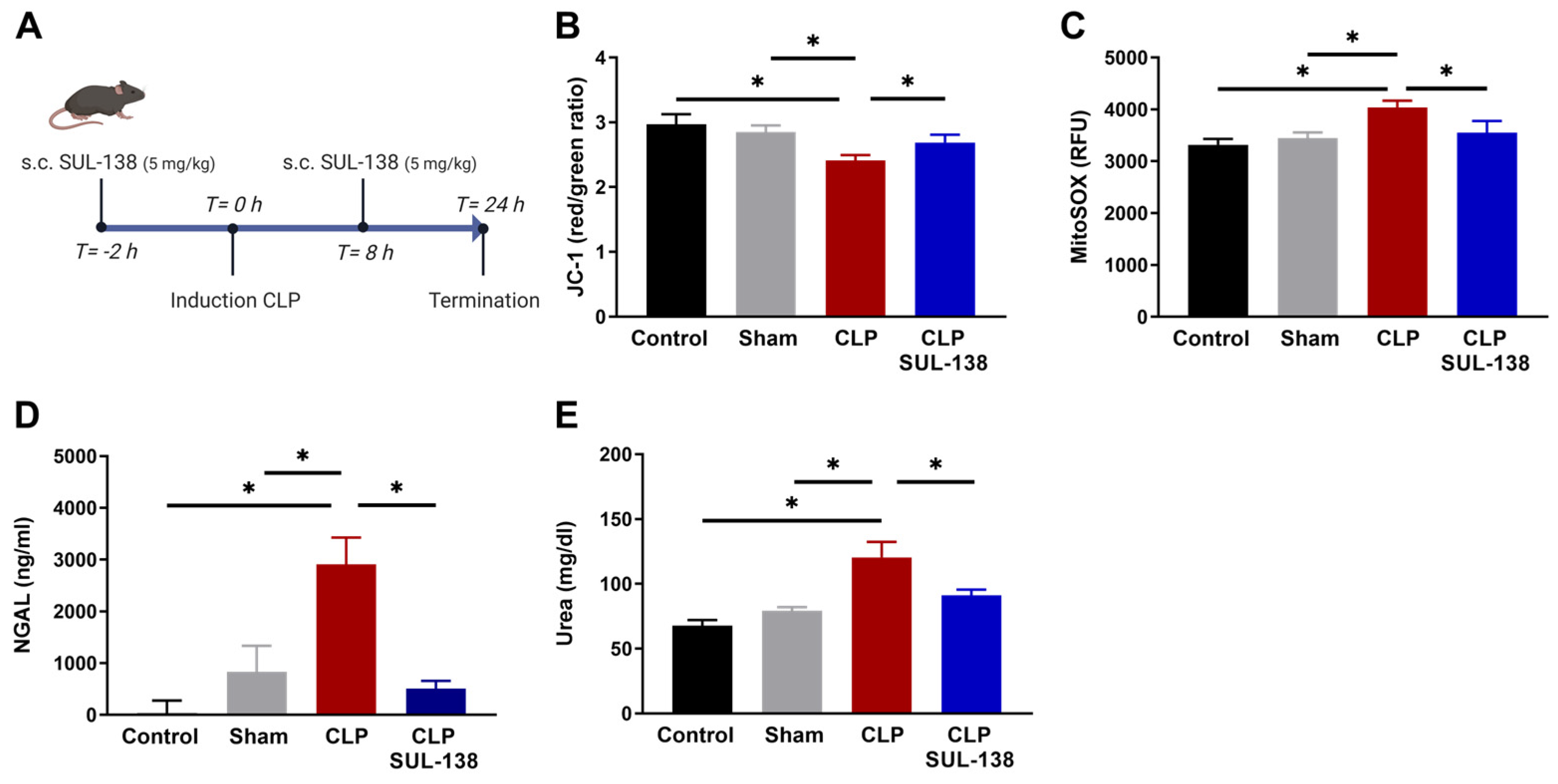
2.3. Effects of SUL-138 on Mitochondrial Function in the Kidney of Septic Mice
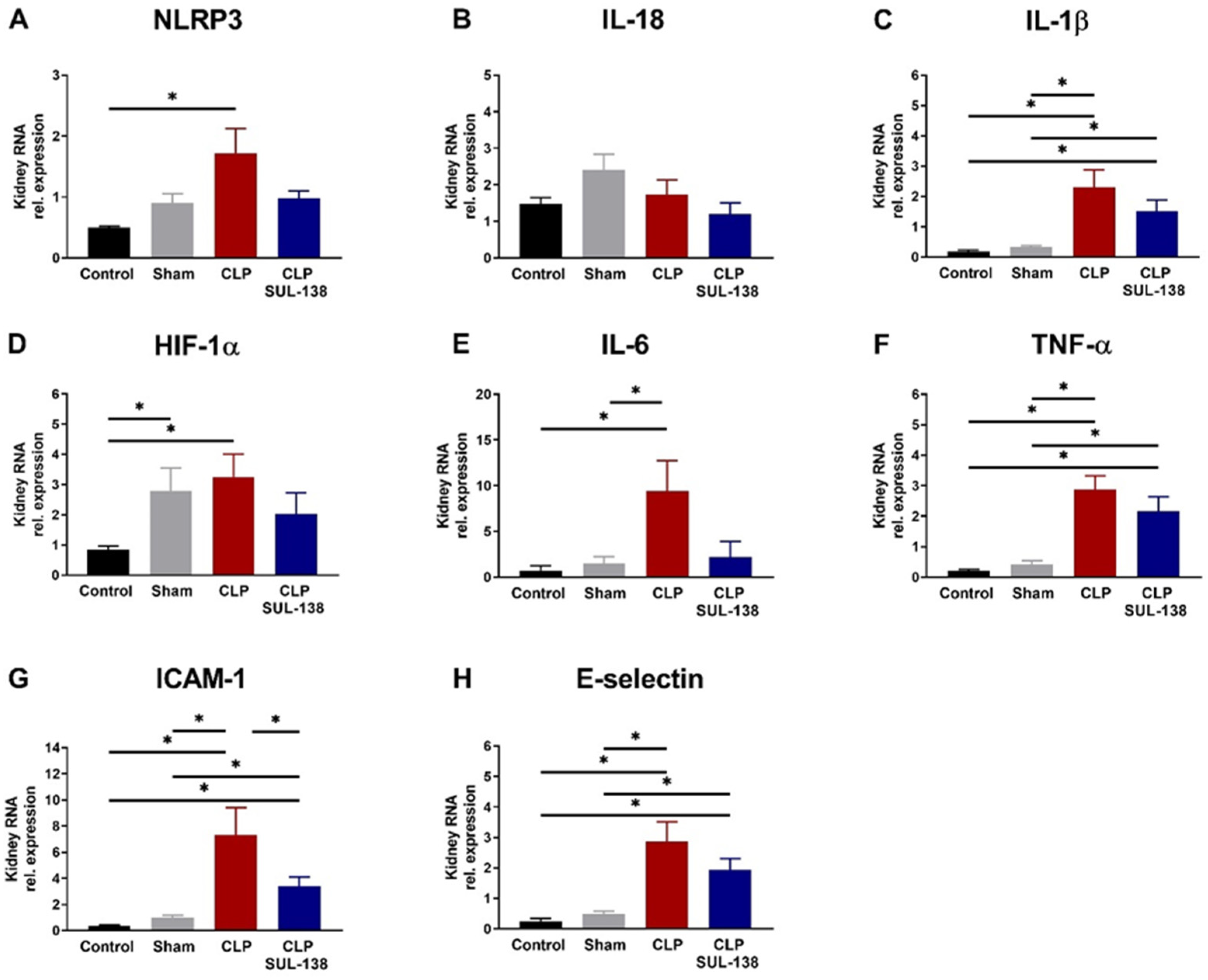
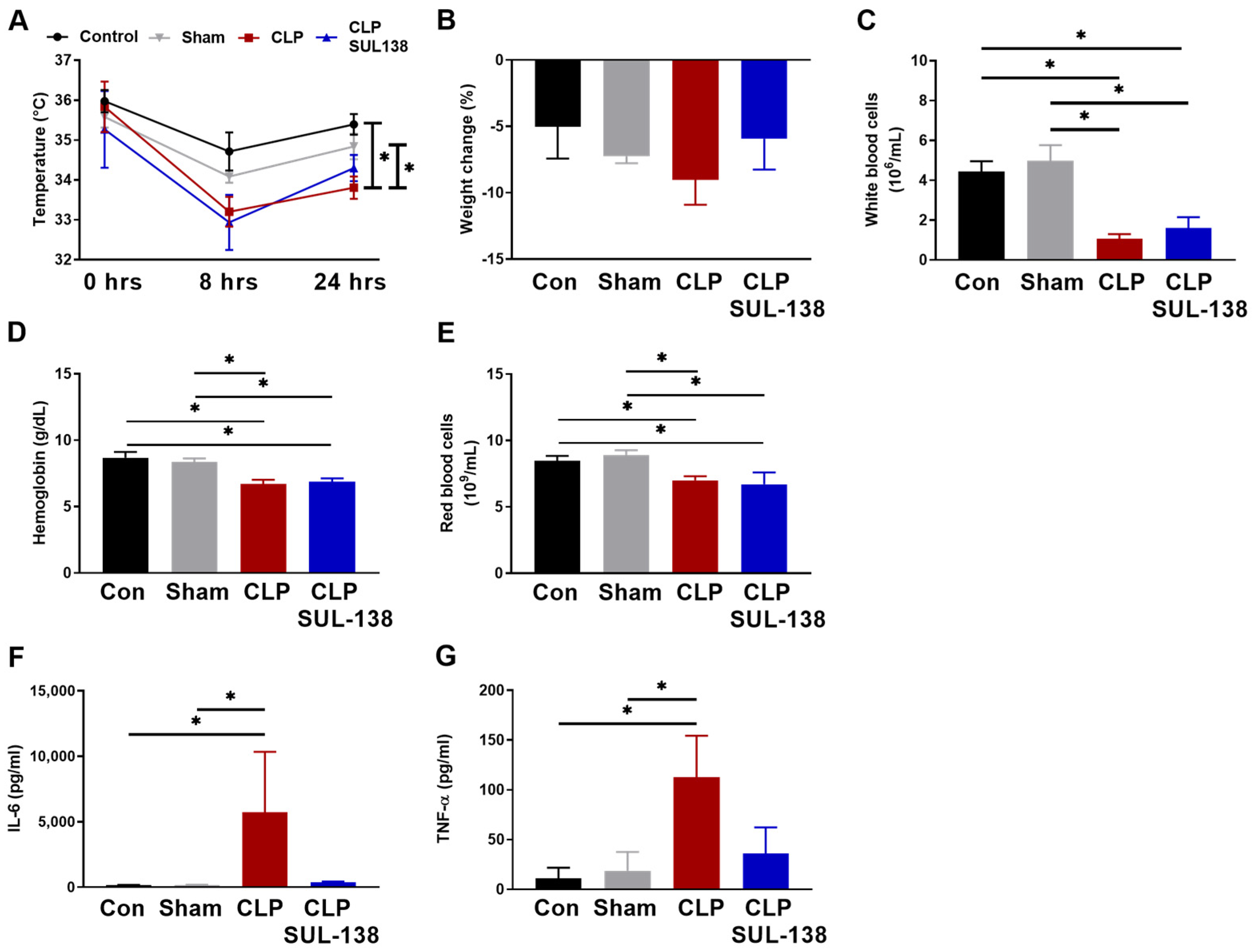
2.4. Effects of SUL-138 on Renal and Systemic Inflammation of Septic Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Endothelial Cell Experimental Setup
4.3. Animal Experimental Setup
4.4. Mitochondria Isolation from the Kidney
4.5. Mitochondrial Membrane Potential
4.6. Mitochondrial Oxidative Stress
4.7. Mitochondrial Oxygen Consumption
4.8. Cell Survival
4.9. Plasma Cytokine Measurement
4.10. Statistical Analysis and Data Presentation
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Sakr, Y.; Jaschinski, U.; Wittebole, X.; Szakmany, T.; Lipman, J.; Namendys-Silva, S.A.; Martin-Loeches, I.; Leone, M.; Lupu, M.N.; Vincent, J.L.; et al. Sepsis in Intensive Care Unit Patients: Worldwide Data From the Intensive Care over Nations Audit. In Open Forum Infectious Diseases; Oxford University Press: Oxford, MS, USA, 2018; Volume 5, p. ofy313. [Google Scholar]
- Hoste, E.A.; Clermont, G.; Kersten, A.; Venkataraman, R.; Angus, D.C.; De Bacquer, D.; Kellum, J.A. RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: A cohort analysis. Crit. Care 2006, 10, R73. [Google Scholar] [CrossRef] [Green Version]
- Uchino, S.; Bellomo, R.; Goldsmith, D.; Bates, S.; Ronco, C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit. Care Med. 2006, 34, 1913–1917. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Alobaidi, R.; Basu, R.K.; Goldstein, S.L.; Bagshaw, S.M. Sepsis-associated acute kidney injury. Semin. Nephrol. 2015, 35, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, J.; Muriel-Bombin, A.; Sagredo, V.; Taboada, F.; Gandia, F.; Tamayo, L.; Collado, J.; Garcia-Labattut, A.; Carriedo, D.; Valledor, M.; et al. Incidence, organ dysfunction and mortality in severe sepsis: A Spanish multicentre study. Crit. Care 2008, 12, R158. [Google Scholar] [CrossRef] [Green Version]
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundler, K.; Angstwurm, M.; Hilge, R.; Baumann, P.; Annecke, T.; Crispin, A.; Sohn, H.Y.; Massberg, S.; Kraemer, B.F. Platelet mitochondrial membrane depolarization reflects disease severity in patients with sepsis and correlates with clinical outcome. Crit. Care 2014, 18, R31. [Google Scholar] [CrossRef] [Green Version]
- Pool, R.; Gomez, H.; Kellum, J.A. Mechanisms of Organ Dysfunction in Sepsis. Crit. Care Clin. 2018, 34, 63–80. [Google Scholar] [CrossRef]
- Zhang, H.X.; Du, J.M.; Ding, Z.N.; Zhu, X.Y.; Jiang, L.; Liu, Y.J. Hydrogen sulfide prevents diaphragm weakness in cecal ligation puncture-induced sepsis by preservation of mitochondrial function. Am. J. Transl. Res. 2017, 9, 3270–3281. [Google Scholar] [PubMed]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted anti-oxidant mitigates injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, D.A.; Thottakam, B.M.; Webster, N.R.; Murphy, M.P.; Galley, H.F. The mitochondria-targeted anti-oxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free. Radic. Biol. Med. 2008, 45, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203. [Google Scholar] [CrossRef]
- Hajmousa, G.; Vogelaar, P.; Brouwer, L.A.; van der Graaf, A.C.; Henning, R.H.; Krenning, G. The 6-chromanol derivate SUL-109 enables prolonged hypothermic storage of adipose tissue-derived stem cells. Biomaterials 2017, 119, 43–52. [Google Scholar] [CrossRef]
- Vogelaar, P.C.; Roorda, M.; de Vrij, E.L.; Houwertjes, M.C.; Goris, M.; Bouma, H.; van der Graaf, A.C.; Krenning, G.; Henning, R.H. The 6-hydroxychromanol derivative SUL-109 ameliorates renal injury after deep hypothermia and rewarming in rats. Nephrol. Dial. Transplant. 2018, 33, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Vogelaar, P.C.; Nakladal, D.; Swart, D.H.; Tkacikova, L.; Tkacikova, S.; van der Graaf, A.C.; Henning, R.H.; Krenning, G. Towards prevention of ischemia-reperfusion kidney injury: Pre-clinical evaluation of 6-chromanol derivatives and the lead compound SUL-138. Eur. J. Pharm. Sci. 2022, 168, 106033. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascon, G.A.; Hernandez, G.; Murray, P.; De Backer, D.; Workgroup, A.X. The Endothelium in Sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Star, B.S.; Boahen, C.K.; van der Slikke, E.C.; Quinten, V.M.; Ter Maaten, J.C.; Henning, R.H.; Kumar, V.; Bouma, H.R. Plasma proteomic characterization of the development of acute kidney injury in early sepsis patients. Sci. Rep. 2022, 12, 19705. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amalakuhan, B.; Habib, S.A.; Mangat, M.; Reyes, L.F.; Rodriguez, A.H.; Hinojosa, C.A.; Soni, N.J.; Gilley, R.P.; Bustamante, C.A.; Anzueto, A.; et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 2016, 88, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, Y.; Ueki, T.; Hata, M.; Iwasawa, K.; Tsuruga, E.; Kojima, H.; Ishikawa, H.; Yoshida, S. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J. Histochem. Cytochem. 2008, 56, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Deng, Z.; Sun, M.; Zhang, W.; Yang, Y.; Zeng, Z.; Wu, J.; Zhang, Q.; Liu, Y.; Chen, Z.; et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation. Lab. Investig. 2020, 100, 643–656. [Google Scholar] [CrossRef]
- Dolmatova, E.V.; Wang, K.; Mandavilli, R.; Griendling, K.K. The effects of sepsis on endothelium and clinical implications. Cardiovasc. Res. 2021, 117, 60–73. [Google Scholar] [CrossRef]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef]
- Oliveira, T.S.; Santos, A.T.; Andrade, C.B.V.; Silva, J.D.; Blanco, N.; Rocha, N.N.; Woyames, J.; Silva, P.L.; Rocco, P.R.M.; da-Silva, W.S.; et al. Sepsis Disrupts Mitochondrial Function and Diaphragm Morphology. Front. Physiol. 2021, 12, 704044. [Google Scholar] [CrossRef]
- Gao, Y.; Zeng, Z.; Li, T.; Xu, S.; Wang, X.; Chen, Z.; Lin, C. Polydatin Inhibits Mitochondrial Dysfunction in the Renal Tubular Epithelial Cells of a Rat Model of Sepsis-Induced Acute Kidney Injury. Anesth. Analg. 2015, 121, 1251–1260. [Google Scholar] [CrossRef]
- Lv, D.; Luo, M.; Yan, J.; Yang, X.; Luo, S. Protective Effect of Sirtuin 3 on CLP-Induced Endothelial Dysfunction of Early Sepsis by Inhibiting NF-kappaB and NLRP3 Signaling Pathways. Inflammation 2021, 44, 1782–1792. [Google Scholar] [CrossRef]
- Peng, Z.Y.; Wang, H.Z.; Srisawat, N.; Wen, X.; Rimmele, T.; Bishop, J.; Singbartl, K.; Murugan, R.; Kellum, J.A. Bactericidal antibiotics temporarily increase inflammation and worsen acute kidney injury in experimental sepsis. Crit. Care Med. 2012, 40, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craciun, F.L.; Iskander, K.N.; Chiswick, E.L.; Stepien, D.M.; Henderson, J.M.; Remick, D.G. Early murine polymicrobial sepsis predominantly causes renal injury. Shock 2014, 41, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, A. Sepsis-induced acute kidney injury. Indian J. Crit. Care Med. 2010, 14, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, Y.W.; Yao, Y.M. Potential therapy strategy: Targeting mitochondrial dysfunction in sepsis. Mil. Med. Res. 2018, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, B.; Yavari, R.; Badalzadeh, R.; Mahmoodpoor, A. An Overview on Mitochondrial-Based Therapies in Sepsis-Related Myocardial Dysfunction: Mitochondrial Transplantation as a Promising Approach. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 3277274. [Google Scholar] [CrossRef]
- de Veij Mestdagh, C.F.; Koopmans, F.; Breiter, J.C.; Timmerman, J.A.; Vogelaar, P.C.; Krenning, G.; Mansvelder, H.D.; Smit, A.B.; Henning, R.H.; van Kesteren, R.E. The hibernation-derived compound SUL-138 shifts the mitochondrial proteome towards fatty acid metabolism and prevents cognitive decline and amyloid plaque formation in an Alzheimer’s disease mouse model. Alzheimer’s Res. Ther. 2022, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Piel, D.A.; Gruber, P.J.; Weinheimer, C.J.; Courtois, M.R.; Robertson, C.M.; Coopersmith, C.M.; Deutschman, C.S.; Levy, R.J. Mitochondrial resuscitation with exogenous cytochrome c in the septic heart. Crit. Care Med. 2007, 35, 2120–2127. [Google Scholar] [CrossRef]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Star, B.S.; van der Slikke, E.C.; van Buiten, A.; Henning, R.H.; Bouma, H.R. The Novel Compound SUL-138 Counteracts Endothelial Cell and Kidney Dysfunction in Sepsis by Preserving Mitochondrial Function. Int. J. Mol. Sci. 2023, 24, 6330. https://doi.org/10.3390/ijms24076330
Star BS, van der Slikke EC, van Buiten A, Henning RH, Bouma HR. The Novel Compound SUL-138 Counteracts Endothelial Cell and Kidney Dysfunction in Sepsis by Preserving Mitochondrial Function. International Journal of Molecular Sciences. 2023; 24(7):6330. https://doi.org/10.3390/ijms24076330
Chicago/Turabian StyleStar, Bastiaan S., Elisabeth C. van der Slikke, Azuwerus van Buiten, Robert H. Henning, and Hjalmar R. Bouma. 2023. "The Novel Compound SUL-138 Counteracts Endothelial Cell and Kidney Dysfunction in Sepsis by Preserving Mitochondrial Function" International Journal of Molecular Sciences 24, no. 7: 6330. https://doi.org/10.3390/ijms24076330