
Deciphering the Mechanism of Tolerance to Apple Replant Disease Using a Genetic Mapping Approach in a Malling 9 × M. × robusta 5 Population Identifies SNP Markers Linked to Candidate Genes


Abstract
:1. Introduction
2. Results
2.1. tGBS SNP Identification
2.2. High-Density Linkage Map Construction
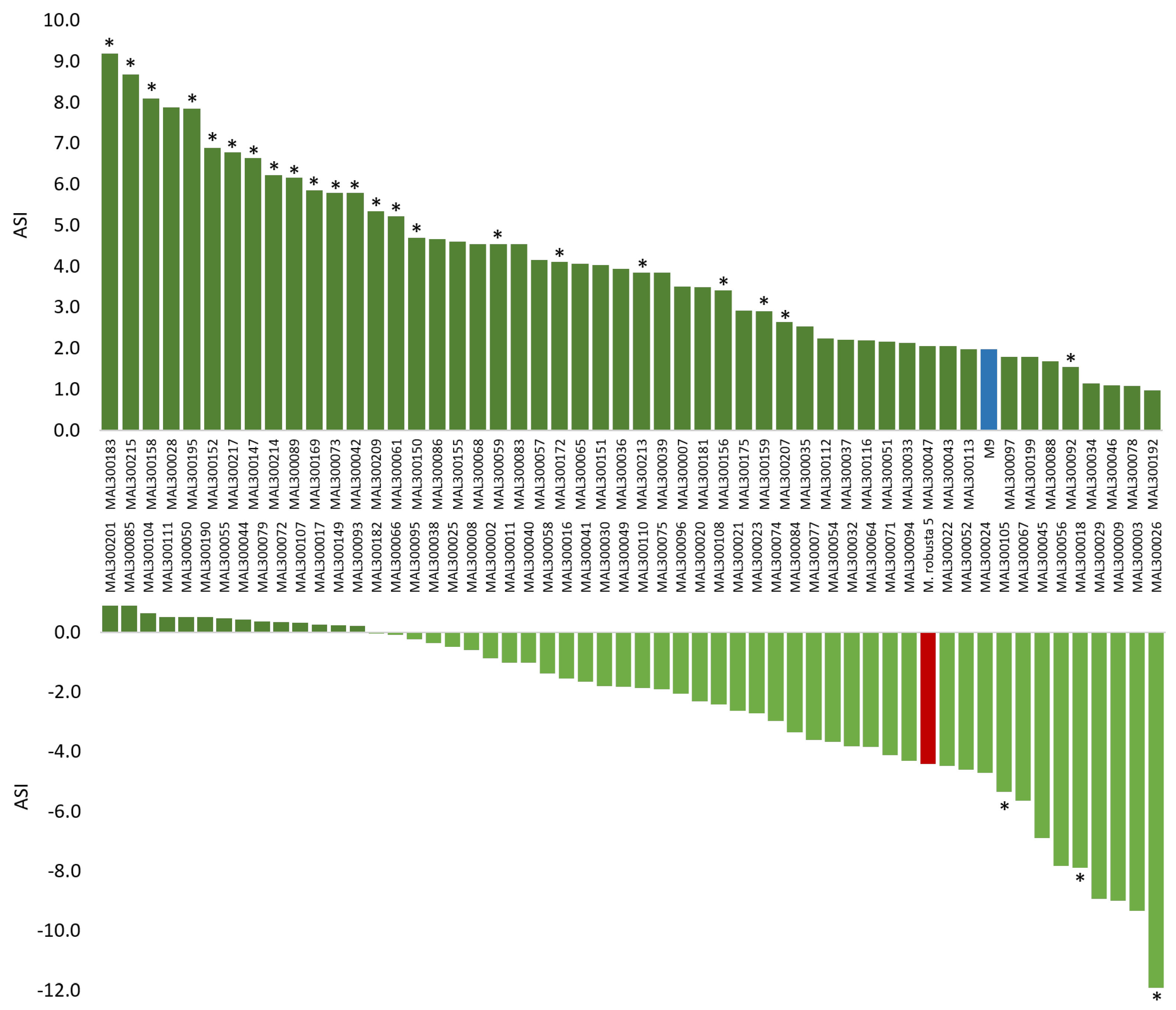
2.3. Phenotypic Analysis
2.4. Marker–Phenotype Association Analyses
2.5. Identification of Potential Candidate Genes in the Malus domestica Genome
3. Discussion
3.1. Construction of a High-Density Genetic Map
3.2. Marker–Phenotype Association Analysis
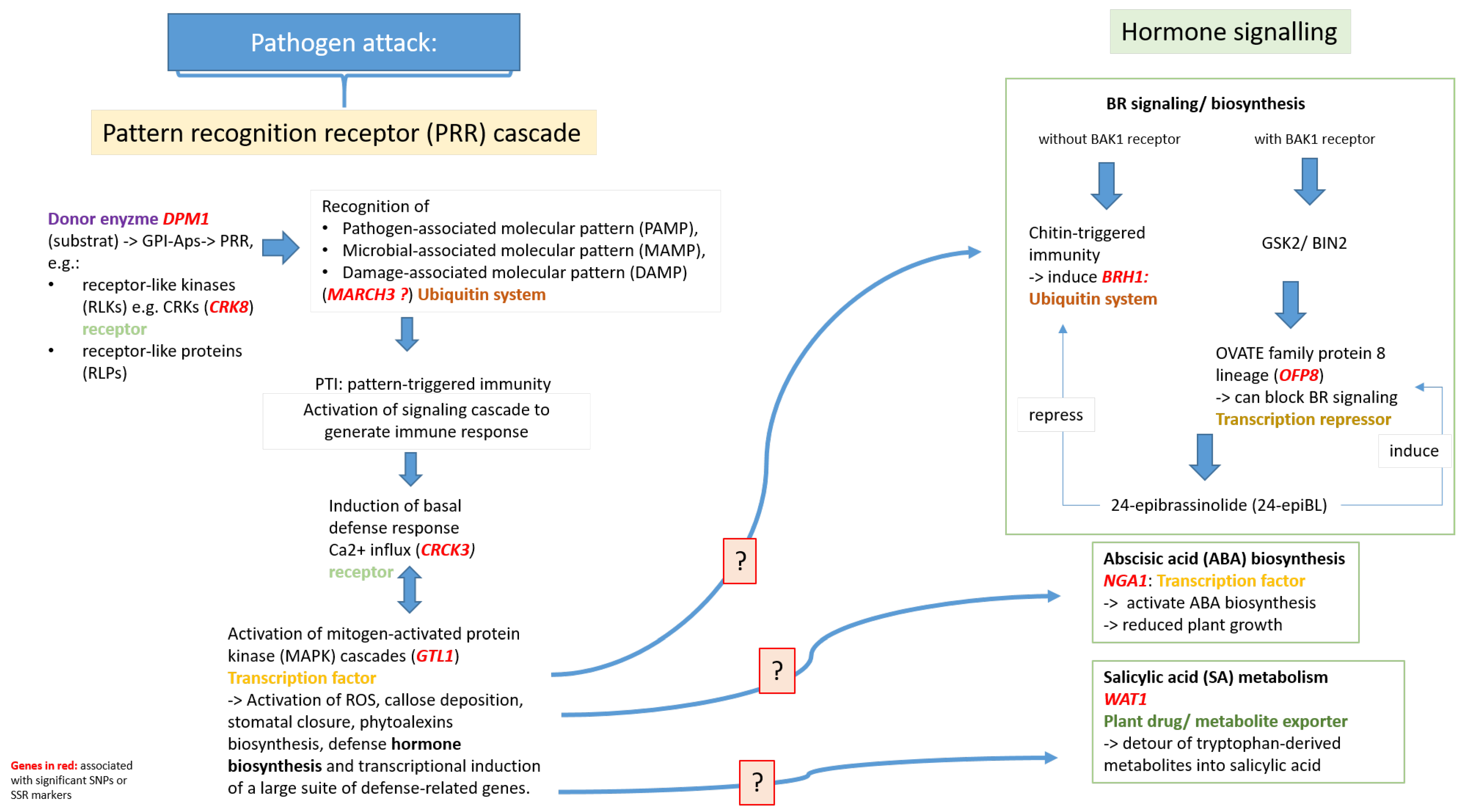
3.3. Candidate Genes
3.3.1. Candidate Genes Involved in Plant Innate Immunity
3.3.2. Candidate Genes Involved in Hormone Synthesis
3.3.3. Salicylic Acid (SA) Metabolism
3.3.4. Abscisic Acid (ABA) Biosynthesis
3.3.5. Brassinosteroid Synthesis
4. Materials and Methods
4.1. Plant Material
4.2. DNA Extraction and SSR Genotyping
4.3. Genotyping-by-Sequencing
4.3.1. Trimming and Alignment of Sequence Reads
4.3.2. SNP Discovery
4.4. Construction of Genetic Maps
4.5. Phenotypic Evaluation and Marker–Phenotype Association Analyses
4.6. Identification of Potential Candidate Genes Linked to Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yim, B.; Nitt, H.; Wrede, A.; Jacquiod, S.; Sorensen, S.J.; Winkelmann, T.; Smalla, K. Effects of soil pre-treatment with basamid (R) granules, Brassica juncea, Raphanus sativus, and Tagetes patula on bacterial and fungal communities at two apple replant disease sites. Front. Microbiol. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed]
- Grunewaldt-Stocker, G.; Popp, C.; Wamhoff, D.; Maiss, E.; Winkelmann, T. Microscopic evidence of Nectriaceae and other microbes in infected fine root tissue of replant diseased apple plants. Eur. J. Hortic. Sci. 2021, 86, 29–40. [Google Scholar] [CrossRef]
- Winkelmann, T.; Smalla, K.; Amelung, W.; Baab, G.; Grunewaldt-Stöcker, G.; Kanfra, X.; Meyhöfer, R.; Reim, S.; Schmitz, M.; Vetterlein, D.; et al. Apple Replant Disease: Causes and Mitigation Strategies. Curr. Issues Mol. Biol. 2019, 30, 89–106. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, M.; Manici, L.M. Apple replant disease: Role of microbial ecology in cause and control. Annu. Rev. Phytopathol. 2012, 50, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M. Elucidation of the microbial complex having a causal role in the development of apple replant disease in Washington. Phytopathology 1998, 88, 930–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewaldt-Stöcker, G.; Mahnkopp, F.; Popp, C.; Maiss, E.; Winkelmann, T. Diagnosis of apple replant disease (ARD): Microscopic evidence of early symptoms in fine roots of different apple rootstock genotypes. Sci. Hortic. 2019, 243, 583–594. [Google Scholar] [CrossRef]
- Popp, C.; Grunewaldt-Stöcker, G.; Maiss, E. A soil-free method for assessing pathogenicity of fungal isolates from apple roots. J. Plant Dis. Protect. 2019, 126, 329–341. [Google Scholar] [CrossRef]
- Popp, C.; Wamhoff, D.; Winkelmann, T.; Maiss, E.; Grunewaldt-Stöcker, G. Molecular identification of Nectriaceae in infections of apple replant disease affected roots collected by Harris Uni-Core punching or laser microdissection. J. Plant Dis. Protect. 2020, 127, 571–582. [Google Scholar] [CrossRef]
- Mahnkopp-Dirks, F.; Radl, V.; Kublik, S.; Gschwendtner, S.; Schloter, M.; Winkelmann, T. Molecular barcoding reveals the genus streptomyces as associated root endophytes of apple (Malus domestica) plants grown in soils affected by Apple Replant Disease. Phytobiomes J. 2021, 5, 177–189. [Google Scholar] [CrossRef]
- Radl, V.; Winkler, J.B.; Kublik, S.; Yang, L.; Winkelmann, T.; Vestergaard, G.; Schröder, P.; Schloter, M. Reduced microbial potential for the degradation of phenolic compounds in the rhizosphere of apple plantlets grown in soils affected by replant disease. Environ. Microbiome 2019, 14, 8. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.N.; Jiang, W.T.; Zhang, R.; Chen, R.; Chen, X.S.; Yin, C.M.; Mao, Z.Q. Discovery of Fusarium proliferatum f. sp. Malus domestica Causing Apple Replant Disease in China. Plant Dis. 2022, 106, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinfelder, M.M.; Merwin, I.A.; Fazio, G.; Robinson, T. Resistant rootstocks, preplant compost amendments, soil fumigation, and row repositioning for managing apple replant disease. HortScience 2004, 39, 841. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Merwin, I.A.; Abawi, G.S.; Thies, J.E. Soil fumigation and compost amendment alter soil microbial community composition but do not improve tree growth or yield in an apple replant site. Soil Biol. Biochem. 2006, 38, 587–599. [Google Scholar] [CrossRef]
- Reim, S.; Rohr, A.-D.; Winkelmann, T.; Weiß, S.; Liu, B.; Beerhues, L.; Schmitz, M.; Hanke, M.-V.; Flachowsky, H. Genes involved in stress response and especially in phytoalexin biosynthesis are upregulated in four malus genotypes in response to Apple Replant Disease. Front. Plant Sci. 2020, 10, 1724. [Google Scholar] [CrossRef]
- Rohr, A.-D.; Schimmel, J.; Liu, B.; Beerhues, L.; Guggenberger, G.; Winkelmann, T. Identification and validation of early genetic biomarkers for apple replant disease. PLoS ONE 2020, 15, e0238876. [Google Scholar] [CrossRef]
- Rohr, A.-D.; Schimmel, J.; Liu, B.; Beerhues, L.; Guggenberger, G.; Winkelmann, T. Correction: Identification and validation of early genetic biomarkers for apple replant disease. PLoS ONE 2022, 17, e0272900. [Google Scholar] [CrossRef] [PubMed]
- Weiß, S.; Bartsch, M.; Winkelmann, T. Transcriptomic analysis of molecular responses in Malus domestica ’M26’ roots affected by apple replant disease. Plant Mol. Biol. 2017, 94, 303–318. [Google Scholar] [CrossRef]
- Zhu, Y.; Fazio, G.; Mazzola, M. Elucidating the molecular responses of apple rootstock resistant to ARD pathogens: Challenges and opportunities for development of genomics-assisted breeding tools. Hortic. Res. 2014, 1, 14043. [Google Scholar] [CrossRef] [Green Version]
- Chizzali, C.; Beerhues, L. Phytoalexins of the Pyrinae: Biphenyls and dibenzofurans. Beilstein J. Org. Chem. 2012, 8, 613–620. [Google Scholar] [CrossRef]
- Leisso, R.; Rudell, D.; Mazzola, M. Targeted metabolic profiling indicates apple rootstock genotype-specific differences in primary and secondary metabolite production and validate quantitative contribution from vegetative growth. Front. Plant Sci. 2018, 9, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Lv, J.; Fazio, G.; Mazzola, M.; Zhu, Y. Transcriptional regulation of ethylene and jasmonate mediated defense response in apple (Malus domestica) root during Pythium ultimum infection. Hortic. Res. 2014, 1, 14053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Lee, J.; Rudell, D.; Evans, K.; Zhu, Y. Transcriptional regulation of auxin metabolism and ethylene biosynthesis activation during apple (Malus × domestica) fruit maturation. J. Plant Growth Regul. 2016, 35, 655–666. [Google Scholar] [CrossRef]
- Weiß, S.; Winkelmann, T. Transcriptome profiling in leaves representing aboveground parts of apple replant disease affected Malus domestica ’M26’ plants. Sci. Hortic. 2017, 222, 111–125. [Google Scholar] [CrossRef]
- Weiß, S.; Liu, B.; Reckwell, D.; Beerhues, L.; Winkelmann, T. Impaired defense reactions in apple replant disease-affected roots of Malus domestica ’M26’. Tree Physiol. 2017, 37, 1672–1685. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.-N.; Li, M.; Wang, W.-J.; Cao, J.; Wang, Z.; Zhu, K.-M.; Yang, Y.-H.; Li, Y.-L.; Tan, X.-L. Advances in plant GDSL lipases: From sequences to functional mechanisms. Acta Physiol. Plant 2019, 41, 151. [Google Scholar] [CrossRef]
- Zhu, Y.; Shao, J.; Zhou, Z.; Davis, R.E. Genotype-specific suppression of multiple defense pathways in apple root during infection by Pythium ultimum. Hortic. Res. 2019, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Reim, S.; Winkelmann, T.; Cestaro, A.; Rohr, A.-D.; Flachowsky, H. Identification of candidate genes associated with tolerance to apple replant disease by genome-wide transcriptome analysis. Front. Microbiol. 2022, 13, 888908. [Google Scholar] [CrossRef]
- Emeriewen, O.F.; Richter, K.; Berner, T.; Keilwagen, J.; Schnable, P.S.; Malnoy, M.; Peil, A. Construction of a dense genetic map of the Malus fusca fire blight resistant accession MAL0045 using tunable genotyping-by-sequencing SNPs and microsatellites. Sci. Rep. 2020, 10, 16358. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.M.; Brown, P.; Cooke, T.F.; Cann, S.; Costa, F.; Bustamante, C.; Velasco, R.; Troggio, M.; Myles, S. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 Genes Genomes Genet. 2014, 4, 1681–1687. [Google Scholar] [CrossRef] [Green Version]
- Reim, S.; Siewert, C.; Winkelmann, T.; Wöhner, T.; Hanke, M.V.; Flachowsky, H. Evaluation of Malus genetic resources for tolerance to apple replant disease (ARD). Sci. Hortic. 2019, 256, 108517. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. MapQTL5, Software for the Mapping of Quantitative Trait Loci in Experimental Populations; Wageningen University & Research: Wageningen, The Netherlands; Kyazma, B.V.: Wageningen, The Netherlands, 2004. [Google Scholar]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Simon, M.; Lehndorff, E.; Wrede, A.; Amelung, W. In-field heterogeneity of apple replant disease: Relations to abiotic soil properties. Sci. Hortic. 2020, 259, 108809. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Jadid, N.; Mialoundama, A.S.; Heintz, D.; Ayoub, D.; Erhardt, M.; Mutterer, J.; Meyer, D.; Alioua, A.; van Dorsselaer, A.; Rahier, A.; et al. DOLICHOL PHOSPHATE MANNOSE SYNTHASE1 mediates the biogenesis of isoprenyl-linked glycans and influences development, stress response, and ammonium hypersensitivity in Arabidopsis. Plant Cell 2011, 23, 1985–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chai, J. Structural Insights into the Plant Immune Receptors PRRs and NLRs. Plant Physiol 2020, 182, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K. Glycosylphosphatidylinositol-Anchored Proteins in Arabidopsis and One of Their Common Roles in Signaling Transduction. Front. Plant Sci. 2019, 10, 1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, A.; Asif, N.; Pirzada, A.R.; Noureen, A.; Shaukat, J.; Burhan, A.; Zaynab, M.; Ali, E.; Imran, K.; Ameen, A.; et al. Genome wide study of cysteine rich receptor like proteins in Gossypium sp. Sci. Rep. 2022, 12, 4885. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, S.; Shu, H.B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samji, T.; Hong, S.; Means, R.E. The Membrane Associated RING-CH Proteins: A Family of E3 Ligases with Diverse Roles through the Cell. Int. Sch. Res. Not. 2014, 2014, 637295. [Google Scholar] [CrossRef]
- Yi, S.Y.; Lee, M.; Kwon, S.-Y.; Kim, W.T.; Lim, Y.P.; Kang, S.-Y. RING-Type E3 Ubiquitin Ligases AtRDUF1 and AtRDUF2 Positively Regulate the Expression of PR1 Gene and Pattern-Triggered Immunity. Int. J. Mol. Sci. 2022, 23, 14525. [Google Scholar] [CrossRef]
- Thulasi Devendrakumar, K.; Li, X.; Zhang, Y. MAP kinase signalling: Interplays between plant PAMP- and effector-triggered immunity. Cell Mol. Life Sci. 2018, 75, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, Y.; Huang, H.; Gao, M.; Wu, D.; Kong, Q.; Zhang, Y. The NLR protein SUMM2 senses the disruption of an immune signaling MAP kinase cascade via CRCK3. EMBO Rep. 2017, 18, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Völz, R.; Kim, S.-K.; Mi, J.; Mariappan, K.G.; Guo, X.; Bigeard, J.; Alejandro, S.; Pflieger, D.; Rayapuram, N.; Al-Babili, S.; et al. The Trihelix transcription factor GT2-like 1 (GTL1) promotes salicylic acid metabolism, and regulates bacterial-triggered immunity. PLoS Genet. 2018, 14, e1007708. [Google Scholar] [CrossRef] [Green Version]
- Weng, H.; Yoo, C.Y.; Gosney, M.J.; Hasegawa, P.M.; Mickelbart, M.V. Poplar GTL1 is a Ca2+/calmodulin-binding transcription factor that functions in plant water use efficiency and drought tolerance. PLoS ONE 2012, 7, e32925. [Google Scholar] [CrossRef] [Green Version]
- Breuer, C.; Kawamura, A.; Ichikawa, T.; Tominaga-Wada, R.; Wada, T.; Kondou, Y.; Muto, S.; Matsui, M.; Sugimoto, K. The trihelix transcription factor GTL1 regulates ploidy-dependent cell growth in the Arabidopsis trichome. Plant Cell 2009, 21, 2307–2322. [Google Scholar] [CrossRef] [Green Version]
- Vidhyasekaran, P. Plant Hormone Signaling Systems in Plant Innate Immunity; Springer: Dordrecht, The Netherlands, 2015; ISBN 9789401792851. [Google Scholar]
- Ranocha, P.; Denancé, N.; Vanholme, R.; Freydier, A.; Martinez, Y.; Hoffmann, L.; Köhler, L.; Pouzet, C.; Renou, J.-P.; Sundberg, B.; et al. Walls are thin 1 (WAT1), an Arabidopsis homolog of Medicago truncatula NODULIN21, is a tonoplast-localized protein required for secondary wall formation in fibers. Plant J. 2010, 63, 469–483. [Google Scholar] [CrossRef]
- Miedes, E.; Vanholme, R.; Boerjan, W.; Molina, A. The role of the secondary cell wall in plant resistance to pathogens. Front. Plant Sci. 2014, 5, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacete, L.; Mélida, H.; Miedes, E.; Molina, A. Plant cell wall-mediated immunity: Cell wall changes trigger disease resistance responses. Plant J. 2018, 93, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Blanco, C.; Feng, D.X.; Hu, J.; Sánchez-Vallet, A.; Deslandes, L.; Llorente, F.; Berrocal-Lobo, M.; Keller, H.; Barlet, X.; Sánchez-Rodríguez, C.; et al. Impairment of cellulose synthases required for Arabidopsis secondary cell wall formation enhances disease resistance. Plant Cell 2007, 19, 890–903. [Google Scholar] [CrossRef] [Green Version]
- Denancé, N.; Ranocha, P.; Oria, N.; Barlet, X.; Rivière, M.-P.; Yadeta, K.A.; Hoffmann, L.; Perreau, F.; Clément, G.; Maia-Grondard, A.; et al. Arabidopsis wat1 (walls are thin1)-mediated resistance to the bacterial vascular pathogen, Ralstonia solanacearum, is accompanied by cross-regulation of salicylic acid and tryptophan metabolism. Plant J. 2013, 73, 225–239. [Google Scholar] [CrossRef]
- Lee, B.H.; Mai, T.T.; Song, J.T.; Kim, J.H. The Arabidopsis thaliana NGATHA1 transcription factor acts as a promoter of a general differentiation program and a carpel identity factor. J. Plant Biol. 2017, 60, 352–357. [Google Scholar] [CrossRef]
- Sasnauskas, G.; Manakova, E.; Lapėnas, K.; Kauneckaitė, K.; Siksnys, V. DNA recognition by Arabidopsis transcription factors ABI3 and NGA1. FEBS J. 2018, 285, 4041–4059. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.H.; Lee, B.H.; Kim, E.Y.; Seo, Y.S.; Lee, S.; Kim, W.T.; Song, J.T.; Kim, J.H. Overexpression of a Brassica rapa NGATHA gene in Arabidopsis thaliana negatively affects cell proliferation during lateral organ and root growth. Plant Cell Physiol. 2009, 50, 2162–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Wang, S.; Li, Y.; Yuan, J.; Xu, L.; Zhang, T.; Chao, Y.; Han, L. Expression of a NGATHA1 Gene from Medicagotruncatula Delays Flowering Time and Enhances Stress Tolerance. Int. J. Mol. Sci. 2020, 21, 2384. [Google Scholar] [CrossRef] [Green Version]
- Kavi Kishor, P.B.; Tiozon, R.N.; Fernie, A.R.; Sreenivasulu, N. Abscisic acid and its role in the modulation of plant growth, development, and yield stability. Trends Plant Sci. 2022, 27, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shen, W.; He, Y.; Tian, Z.; Li, J. OVATE Family Protein 8 Positively Mediates Brassinosteroid Signaling through Interacting with the GSK3-like Kinase in Rice. PLoS Genet. 2016, 12, e1006118. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Sae-Seaw, J.; Wang, Z.-Y. Brassinosteroid signalling. Development 2013, 140, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, A.L.G.L.; Soares, J.S.; Tavares, R.G.; Righetto, G.; Zullo, M.A.T.; Mandava, N.B.; Menossi, M. Brassinosteroids, the Sixth Class of Phytohormones: A Molecular View from the Discovery to Hormonal Interactions in Plant Development and Stress Adaptation. Int. J. Mol. Sci. 2019, 20, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.-H.; Zhao, Z.-Z.; He, J.-X. Brassinosteroid Signaling in Plant-Microbe Interactions. Int. J. Mol. Sci. 2018, 19, 4091. [Google Scholar] [CrossRef] [Green Version]
- Molnár, G.; Bancoş, S.; Nagy, F.; Szekeres, M. Characterisation of BRH1, a brassinosteroid-responsive RING-H2 gene from Arabidopsis thaliana. Planta 2002, 215, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Kumar, G.B.S.; Khan, M.; Doohan, F.M. Brassinosteroid enhances resistance to fusarium diseases of barley. Phytopathology 2013, 103, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Che, X.; Liang, J.; Wang, S.; Han, L.; Liu, Z.; Chen, H.; Tang, M. Brassinosteroids Benefit Plants Performance by Augmenting Arbuscular Mycorrhizal Symbiosis. Microbiol. Spectr. 2021, 9, e0164521. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, E.; Ge, X.; Gong, Q.; Butt, H.; Zhang, C.; Yang, Z.; Li, F.; Zhang, X. Overexpressed BRH1, a RING finger gene, alters rosette leaf shape in Arabidopsis thaliana. Sci. China Life Sci. 2018, 61, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ooijen, J.W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. Accuracy of mapping quantitative trait loci in autogamous species. Theor. Appl. Genet. 1992, 84, 803–811. [Google Scholar] [CrossRef]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- McNamara, N.P.; Black, H.I.; Beresford, N.A.; Parekh, N.R. Effects of acute gamma irradiation on chemical, physical and biological properties of soils. Appl. Soil Ecol. 2003, 24, 117–132. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. MapQTL, Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species; ScienceOpen, Inc.: Burlington, MA, USA, 2009. [Google Scholar]



{kind=link}
{kind=link}
| ‘M9’ | Mr5 | |||||
|---|---|---|---|---|---|---|
| LG | Length (cM) | SNP Marker (n) | SSR Marker (n) | Length (cM) | SNP Marker (n) | SSR Marker (n) |
| LG1 | 64.83 | 65 | 3 | 59.94 | 94 | 4 |
| LG2 | 82.03 | 109 | 6 | 52.10 | 176 | 5 |
| LG3 | 70.36 | 132 | 5 | 56.29 | 246 | 5 |
| LG4 | 52.95 | 126 | 1 | 66.69 | 114 | 4 |
| LG5 | 80.87 | 131 | 7 | 86.15 | 83 | 7 |
| LG6 | 73.19 | 50 | 3 | 89.61 | 266 | 6 |
| LG7 | 78.81 | 100 | 3 | 63.53 | 104 | 4 |
| LG8 | 56.81 | 122 | 0 | 54.95 | 129 | 0 |
| LG9 | 65.38 | 189 | 3 | 60.56 | 165 | 3 |
| LG10 | 84.35 | 139 | 2 | 72.30 | 178 | 2 |
| LG11 | 94.15 | 94 | 2 | 54.04 | 267 | 3 |
| LG12 | 63.51 | 88 | 2 | 55.21 | 199 | 2 |
| LG13 | 60.51 | 107 | 3 | 62.92 | 322 | 4 |
| LG14 | 65.12 | 127 | 0 | 52.93 | 186 | 1 |
| LG15 | 106.86 | 115 | 5 | 76.33 | 180 | 4 |
| LG16 | 76.87 | 116 | 4 | 75.18 | 149 | 5 |
| LG17 | 77.32 | 124 | 6 | 58.96 | 131 | 5 |
| sum | 1253.92 | 1879 | 55 * | 1097.70 | 2925 | 64 * |
| Marker | LG | Start | End | ‘M9’ | Mr5 | Position 1 | K* 2 | Signif. 3 |
|---|---|---|---|---|---|---|---|---|
| SNP_31883 | 6 | 26,471,091 | 26,470,957 | G | G/T | 52.66 | 8.01 | **** |
| SNP_32246 | 6 | 26,701,927 | 26,701,806 | T | T/A | 52.31 | 7.94 | **** |
| SNP_36646 | 6 | 26,470,806 | 26,470,950 | A | A/G | 52.78 | 8.89 | **** |
| SNP_36649 | 6 | 26,470,844 | 26,470,904 | A | A/C | 52.82 | 8.57 | **** |
| SNP_49388 | 6 | 26,982,732 | 26,982,590 | T | T/C | 52.41 | 8.08 | **** |
| SNP_54395 | 6 | 26,733,503 | 26,733,640 | G | G/T | 52.73 | 8.46 | **** |
| SNP_56130 | 6 | 26,648,239 | 26,648,380 | C | C/G | 52.13 | 8.14 | **** |
| SNP_56298 | 6 | 24,029,075 | 24,028,955 | C | C/G | 56.74 | 8.06 | **** |
| SNP_56970 | 6 | 26,705,196 | 26,705,318 | A | A/T | 52.63 | 8.27 | **** |
| SNP_57966 | 6 | 26,733,715 | 26,733,603 | C | C/T | 52.85 | 8.08 | **** |
| SNP_58944 | 6 | 24,726,042 | 24,726,172 | A | A/T | 55.97 | 8.98 | **** |
| SNP_58943 | 6 | 24,726,042 | 24,726,168 | C | C/T | 55.78 | 8.24 | **** |
| SNP_52341 | 2 | 1,549,119 | 1,548,999 | G | G/A | 8.31 | 8.62 | **** |
| Ch02c02a | 2 | 3,716,747 | 3,716,507 | 178/192 | 0/184 ### | 75.94 #/0 ## | 12.95 | **** |
| Gene ID | LG | Start | End | Marker | Start | End | NCBI | Tair | Symbol | Function |
|---|---|---|---|---|---|---|---|---|---|---|
| MD02G1046400 | 2 | 3,703,811 | 3,705,009 | Ch02c02a | 3,716,747 | 3,716,507 | XP_008386472 | AT5G19650.1 | OFP8 | Transcription repressor OFP8-like [Malus domestica] |
| MD02G1022200 | 2 | 1,548,373 | 1,552,372 | SNP_52341 | 1,549,119 | 1,548,999 | XP_008385188 | AT2G11520.1 | CRCK3 | Calmodulin-binding receptor-like cytoplasmic kinase 3 [Malus domestica] |
| MD06G1103200 | 6 | 24,022,662 | 24,023,312 | SNP_56298 | 24,029,075 | 24,028,955 | XP_008351649 | AT5G41810.2 | LOC103415066 | Uncharacterized protein LOC103415066 [Malus domestica] |
| MD06G1107200 | 6 | 24,688,790 | 24,691,604 | SNP_58943 | 24,726,042 | 24,726,168 | CAN80145 | AT4G23160.1 | CRK8 | Cysteine-rich RLK (RECEPTOR-like protein kinase) 8 |
| 6 | SNP_58944 | 24,726,042 | 24,726,172 | |||||||
| MD06G1123000 | 6 | 26,462,265 | 26,462,819 | SNP_36646 | 26,470,806 | 26,470,950 | XP_008358490 | AT3G61460.1 | BRH1 | E3 ubiquitin-protein ligase RHA2B-like [Malus domestica] |
| 6 | SNP_36649 | 26,470,844 | 26,470,904 | |||||||
| 6 | SNP_31883 | 26,471,091 | 26,470,957 | |||||||
| MD06G1124400 | 6 | 26,645,912 | 26,650,494 | SNP_56130 | 26,648,239 | 26,648,380 | XP_008375464 | AT2G46870.1 | NGA1 | B3 domain-containing protein Os03g0120900-like [Malus domestica] |
| MD06G1124900 | 6 | 26,701,497 | 26,703,758 | SNP_32246 | 26,701,927 | 26,701,806 | XP_008375454 | AT2G45530.1 | MARCH3 | E3 ubiquitin-protein ligase MARCH3 isoform X2 [Malus domestica] |
| MD06G1125000 | 6 | 26,704,481 | 26,707,546 | SNP_56970 | 26,705,196 | 26,705,318 | XP_008375452 | AT1G20575.1 | DPM1 | Dolichol-phosphate mannosyltransferase subunit 1 [Malus domestica] |
| MD06G1125400 | 6 | 26,731,654 | 26,733,668 | SNP_54395 | 26,733,503 | 26,733,640 | XP_008375451 | AT3G30340.1 | WAT1 | WAT1-related protein At3g30340 isoform X2 [Malus domestica] |
| 6 | SNP_57966 | 26,733,715 | 26,733,603 | |||||||
| MD06G1127800 | 6 | 26,980,060 | 26,984,800 | SNP_49388 | 26,982,732 | 26,982,590 | XP_008375438 | AT1G76880.1 | GTL1 | Trihelix transcription factor GTL1 isoform X1 [Malus domestica] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reim, S.; Emeriewen, O.F.; Peil, A.; Flachowsky, H. Deciphering the Mechanism of Tolerance to Apple Replant Disease Using a Genetic Mapping Approach in a Malling 9 × M. × robusta 5 Population Identifies SNP Markers Linked to Candidate Genes. Int. J. Mol. Sci. 2023, 24, 6307. https://doi.org/10.3390/ijms24076307
Reim S, Emeriewen OF, Peil A, Flachowsky H. Deciphering the Mechanism of Tolerance to Apple Replant Disease Using a Genetic Mapping Approach in a Malling 9 × M. × robusta 5 Population Identifies SNP Markers Linked to Candidate Genes. International Journal of Molecular Sciences. 2023; 24(7):6307. https://doi.org/10.3390/ijms24076307
Chicago/Turabian StyleReim, Stefanie, Ofere Francis Emeriewen, Andreas Peil, and Henryk Flachowsky. 2023. "Deciphering the Mechanism of Tolerance to Apple Replant Disease Using a Genetic Mapping Approach in a Malling 9 × M. × robusta 5 Population Identifies SNP Markers Linked to Candidate Genes" International Journal of Molecular Sciences 24, no. 7: 6307. https://doi.org/10.3390/ijms24076307