
The Voltage-Gated Hv1 H+ Channel Is Expressed in Tumor-Infiltrating Myeloid-Derived Suppressor Cells
 , and
, and
Abstract
:
1. Introduction
2. Results
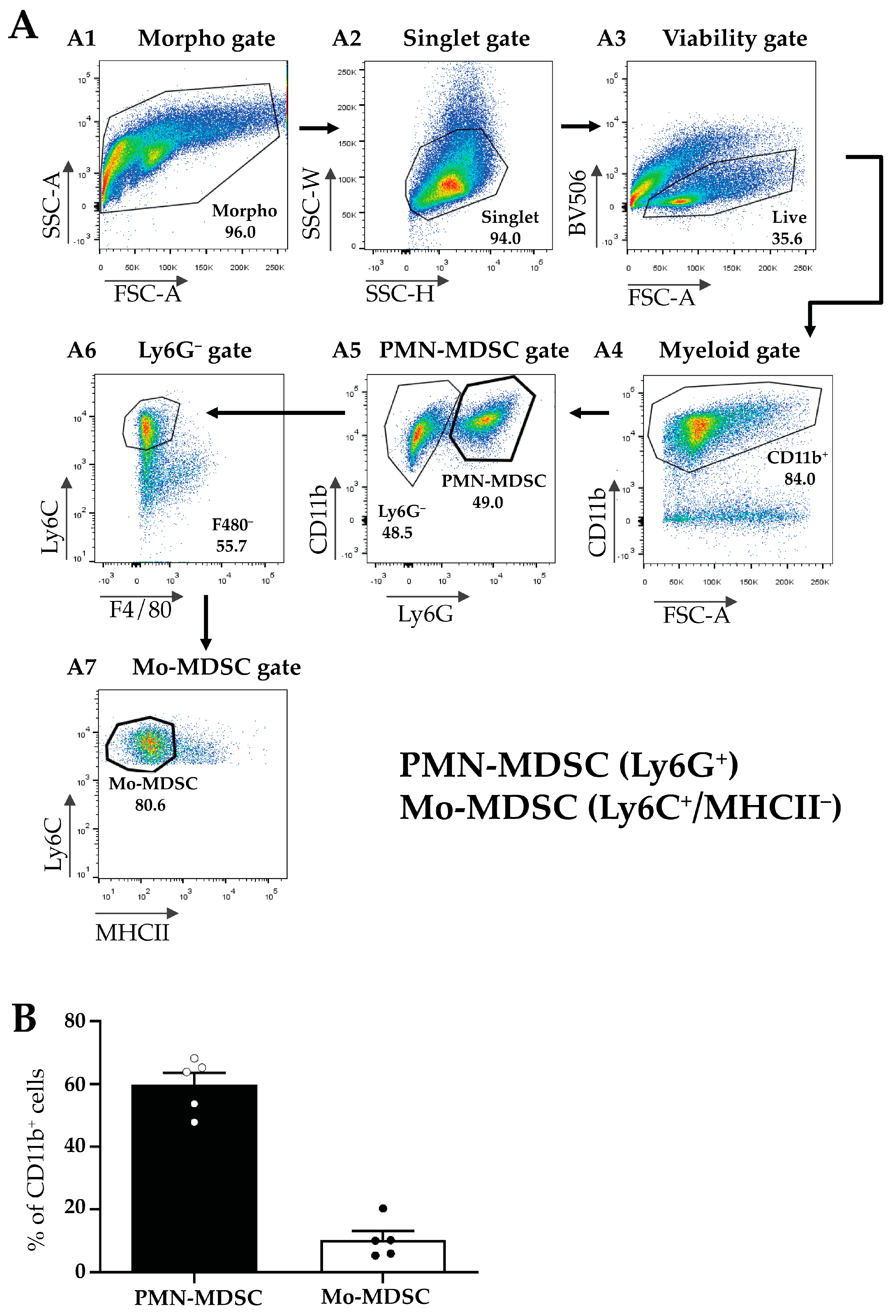
2.1. Identification, Isolation, and Functional Characterization of Tumor-Associated Mo- and PMN-MDCs
2.2. Detection of Hv1 Proton Channel in Tumor Derived MDSCs
2.3. Expression of Hv1 in MDSCs Infiltrating LLC Tumors
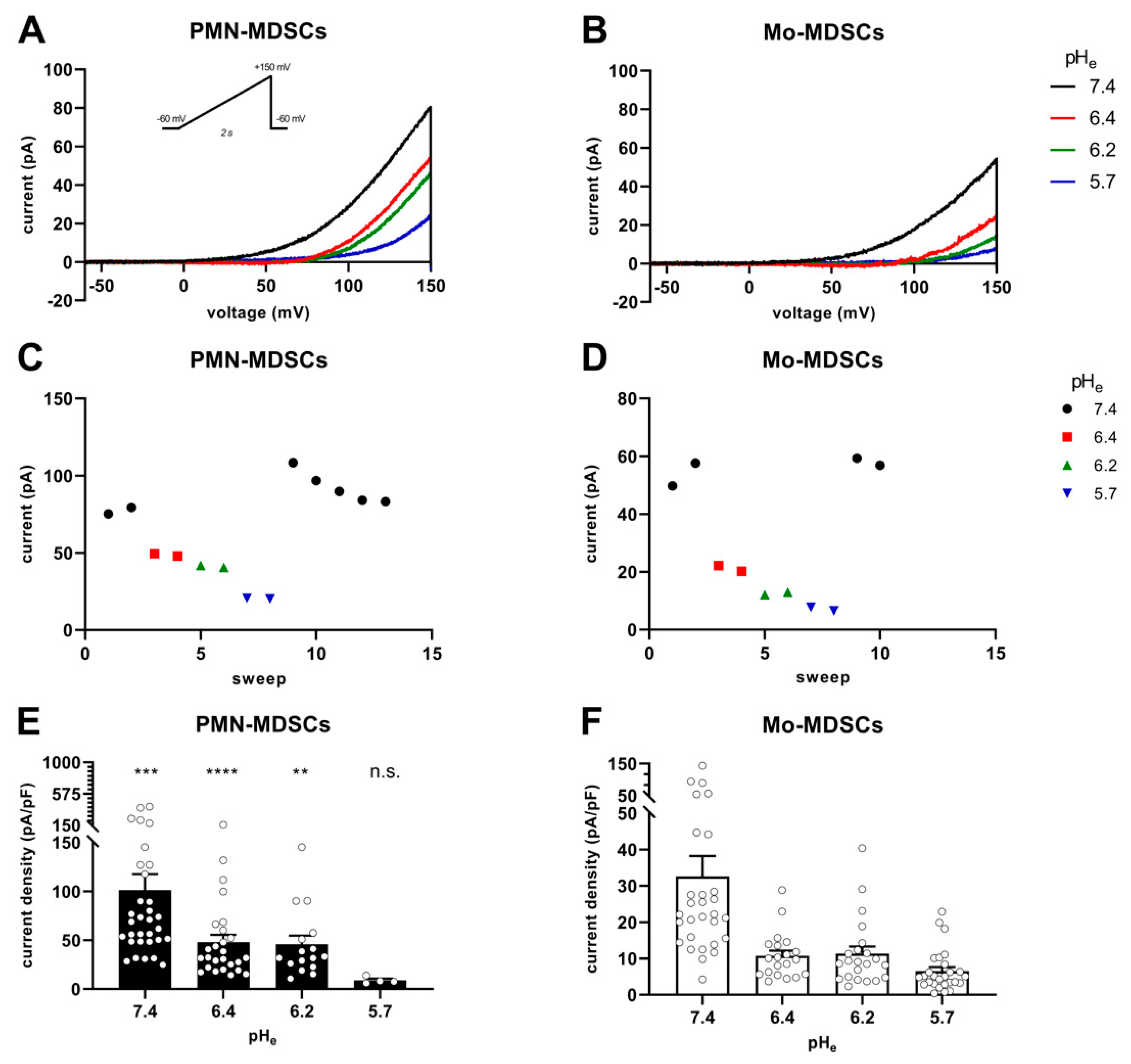
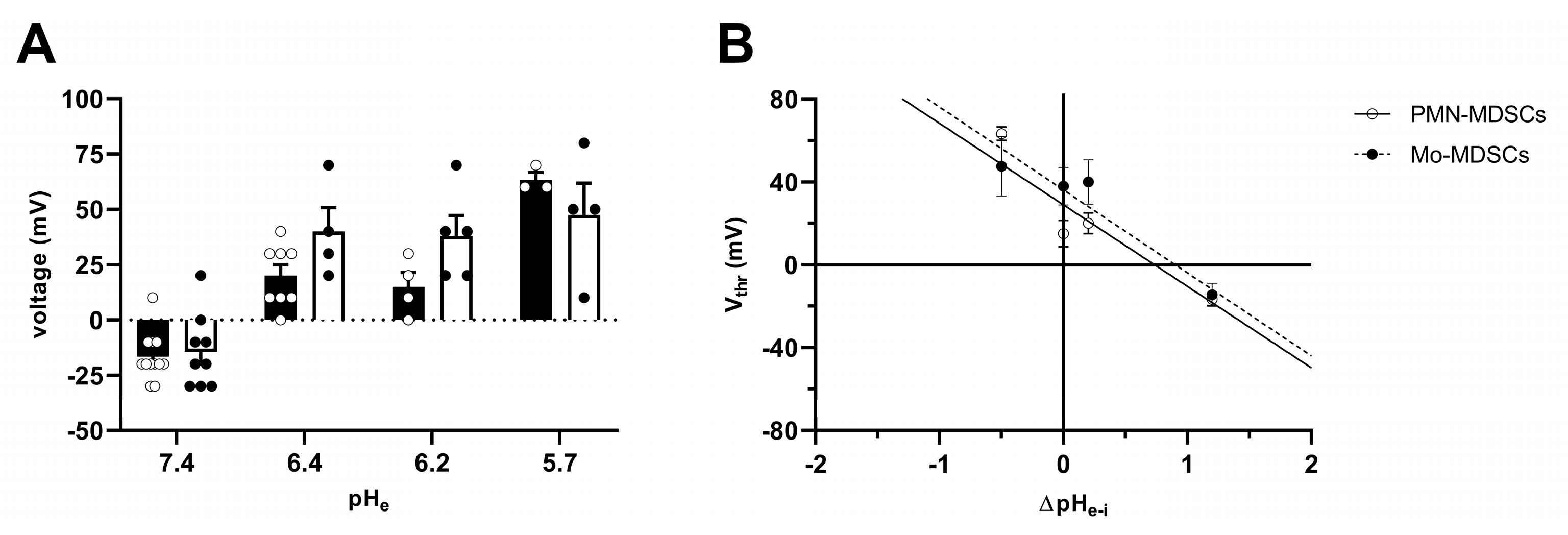
2.4. Ion Currents in MDSCs
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cells
4.3. Tumor Model and Cell Sorting
4.4. Suppression of Polyclonal T Cell Proliferation
4.5. Real Time Quantitative PCR (RT-qPCR)
4.6. Western Blot
4.7. Immunofluorescence
4.8. Electrophysiology and Pharmacology
4.9. Electrophysiology Data Acquisition and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [Green Version]
- De Sanctis, F.; Bronte, V.; Ugel, S. Tumor-Induced Myeloid-Derived Suppressor Cells. Microbiol. Spectr. 2016, 4, 833–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [Green Version]
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; Andre, P.; Rigot, V.; Iovanna, J.; van de Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2019, 10, 3070. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, J.M.; Smith, A.M.; Weinlich, R.; Dillon, C.P.; Qualls, J.E.; Neale, G.; Koss, B.; Kim, Y.; Bronte, V.; Herold, M.J.; et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 2014, 41, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenhals, J.E.; Seyedin, S.N.; Anderson, C.; Brooks, E.D.; Li, Y.R.; Younes, A.I.; Niknam, S.; Li, A.; Barsoumian, H.B.; Cortez, M.A.; et al. Uncovering the immune tumor microenvironment in non-small cell lung cancer to understand response rates to checkpoint blockade and radiation. Transl. Lung Cancer Res. 2017, 6, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Nagaoka, K.; Kobayashi, Y.; Nakagawa, H.; Kakimi, K.; Nakajima, J. Neoantigen Dendritic Cell Vaccination Combined with Anti-CD38 and CpG Elicits Anti-Tumor Immunity against the Immune Checkpoint Therapy-Resistant Murine Lung Cancer Cell Line LLC1. Cancers 2021, 13, 5508. [Google Scholar] [CrossRef]
- Suk Lee, Y.; Davila, E.; Zhang, T.; Milmoe, H.P.; Vogel, S.N.; Bromberg, J.S.; Scalea, J.R. Myeloid-derived suppressor cells are bound and inhibited by anti-thymocyte globulin. Innate Immun. 2019, 25, 46–59. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, Q.; Wang, T.; Lin, H.; Zhan, Q.; Chu, Z.; Huang, R.; Zhou, X.; Liang, X.; Guo, W. MicroRNA expression profiles of granulocytic myeloid-derived suppressor cells from mice bearing Lewis lung carcinoma. Mol. Med. Rep. 2016, 14, 4567–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Wang, H.; Zhuang, X. Myeloid-derived suppressor cells enhance the expression of melanoma-associated antigen A4 in a Lewis lung cancer murine model. Oncol. Lett. 2016, 11, 809–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.Y.; McSharry, M.; Bullock, B.; Nguyen, T.T.; Kwak, J.; Poczobutt, J.M.; Sippel, T.R.; Heasley, L.E.; Weiser-Evans, M.C.; Clambey, E.T.; et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol. Res. 2017, 5, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Feske, S. Ion channelopathies of the immune system. Curr. Opin. Immunol. 2018, 52, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.L.; Nagarkatti, P.S.; Nagarkatti, M. Role of myeloid-derived suppressor cells in amelioration of experimental autoimmune hepatitis following activation of TRPV1 receptors by cannabidiol. PLoS ONE 2011, 6, e18281. [Google Scholar] [CrossRef] [Green Version]
- Alvear-Arias, J.J.; Carrillo, C.; Villar, J.P.; Garcia-Betancourt, R.; Pena-Pichicoi, A.; Fernandez, A.; Fernandez, M.; Carmona, E.M.; Pupo, A.; Neely, A.; et al. Expression of H(v)1 proton channels in myeloid-derived suppressor cells (MDSC) and its potential role in T cell regulation. Proc. Natl. Acad. Sci. USA 2022, 119, e2104453119. [Google Scholar] [CrossRef]
- Tombola, F.; Ulbrich, M.H.; Isacoff, E.Y. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron 2008, 58, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, I.S.; Moran, M.M.; Chong, J.A.; Clapham, D.E. A voltage-gated proton-selective channel lacking the pore domain. Nature 2006, 440, 1213–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCoursey, T.E.; Cherny, V.V.; Zhou, W.; Thomas, L.L. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc. Natl. Acad. Sci. USA 2000, 97, 6885–6889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petheo, G.L.; Orient, A.; Barath, M.; Kovacs, I.; Rethi, B.; Lanyi, A.; Rajki, A.; Rajnavolgyi, E.; Geiszt, M. Molecular and functional characterization of Hv1 proton channel in human granulocytes. PLoS ONE 2010, 5, e14081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okochi, Y.; Sasaki, M.; Iwasaki, H.; Okamura, Y. Voltage-gated proton channel is expressed on phagosomes. Biochem. Biophys. Res. Commun. 2009, 382, 274–279. [Google Scholar] [CrossRef] [PubMed]
- DeCoursey, T.E.; Cherny, V.V.; DeCoursey, A.G.; Xu, W.; Thomas, L.L. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J. Physiol. 2001, 535, 767–781. [Google Scholar] [CrossRef]
- Capasso, M.; Bhamrah, M.K.; Henley, T.; Boyd, R.S.; Langlais, C.; Cain, K.; Dinsdale, D.; Pulford, K.; Khan, M.; Musset, B.; et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 2010, 11, 265–272. [Google Scholar] [CrossRef]
- Montes-Cobos, E.; Huscher, B.; Engler, J.B.; Woo, M.S.; Binkle, L.; Bauer, S.; Kursawe, N.; Moles, M.; Friese, M.A.; Ufer, F. Voltage-Gated Proton Channel Hv1 Controls TLR9 Activation in Plasmacytoid Dendritic Cells. J. Immunol. 2020, 205, 3001–3010. [Google Scholar] [CrossRef]
- Schilling, T.; Gratopp, A.; DeCoursey, T.E.; Eder, C. Voltage-activated proton currents in human lymphocytes. J. Physiol. 2002, 545, 93–105. [Google Scholar] [CrossRef]
- Coe, D.; Poobalasingam, T.; Fu, H.; Bonacina, F.; Wang, G.; Morales, V.; Moregola, A.; Mitro, N.; Cheung, K.C.; Ward, E.J.; et al. Loss of voltage-gated hydrogen channel 1 expression reveals heterogeneous metabolic adaptation to intracellular acidification by T cells. JCI Insight 2022, 7, e147814. [Google Scholar] [CrossRef]
- Fernandez, A.; Pupo, A.; Mena-Ulecia, K.; Gonzalez, C. Pharmacological Modulation of Proton Channel Hv1 in Cancer Therapy: Future Perspectives. Mol. Pharmacol. 2016, 90, 385–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petho, Z.; Najder, K.; Carvalho, T.; McMorrow, R.; Todesca, L.M.; Rugi, M.; Bulk, E.; Chan, A.; Lowik, C.; Reshkin, S.J.; et al. pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers 2020, 12, 2484. [Google Scholar] [CrossRef]
- Asuaje, A.; Smaldini, P.; Martin, P.; Enrique, N.; Orlowski, A.; Aiello, E.A.; Gonzalez Leon, C.; Docena, G.; Milesi, V. The inhibition of voltage-gated H(+) channel (HVCN1) induces acidification of leukemic Jurkat T cells promoting cell death by apoptosis. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Capasso, M. Regulation of immune responses by proton channels. Immunology 2014, 143, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Carmona, M.; Lucas-Ruiz, F.; Gallego-Ortega, A.; Galindo-Romero, C.; Norte-Munoz, M.; Gonzalez-Riquelme, M.J.; Valiente-Soriano, F.J.; Vidal-Sanz, M.; Agudo-Barriuso, M. Ly6c as a New Marker of Mouse Blood Vessels: Qualitative and Quantitative Analyses on Intact and Ischemic Retinas. Int. J. Mol. Sci. 2021, 23, 19. [Google Scholar] [CrossRef]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Lore, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Takagi, M.; Okamura, Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science 2006, 312, 589–592. [Google Scholar] [CrossRef]
- Sokolov, V.S.; Cherny, V.V.; Ayuyan, A.G.; DeCoursey, T.E. Analysis of an electrostatic mechanism for DeltapH dependent gating of the voltage-gated proton channel, H(V)1, supports a contribution of protons to gating charge. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2021, 1862, 148480. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Papp, F.; Mocsar, G.; Kokai, E.; Kovacs, K.; Tajti, G.; Panyi, G. The voltage-gated proton channel hHv1 is functionally expressed in human chorion-derived mesenchymal stem cells. Sci. Rep. 2020, 10, 7100. [Google Scholar] [CrossRef]
- Musset, B.; Decoursey, T. Biophysical properties of the voltage gated proton channel H(V)1. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Kim, I.H.; Tombola, F. Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc. Natl. Acad. Sci. USA 2014, 111, 9971–9976. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Yescas, G.; Cervantes, C.; Cervantes-Rocha, M.A.; Suarez-Delgado, E.; Banaszak, A.T.; Maldonado, E.; Ramsey, I.S.; Rosenbaum, T.; Islas, L.D. Discovery and characterization of H(v)1-type proton channels in reef-building corals. Elife 2021, 10, e69248. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Zhang Md, J.; Zhang Md, L.; Yang Md, Y.; Liu Md, Q.; Ma Md, H.; Huang Md, A.; Zhao Md, Y.; Xia Md, Z.; Liu Md, T.; Wu Md, G. Polymorphonuclear-MDSCs Facilitate Tumor Regrowth After Radiation by Suppressing CD8(+) T Cells. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1533–1546. [Google Scholar] [CrossRef]
- Metzger, P.; Kirchleitner, S.V.; Boehmer, D.F.R.; Horth, C.; Eisele, A.; Ormanns, S.; Gunzer, M.; Lech, M.; Lauber, K.; Endres, S.; et al. Systemic but not MDSC-specific IRF4 deficiency promotes an immunosuppressed tumor microenvironment in a murine pancreatic cancer model. Cancer Immunol. Immunother. 2020, 69, 2101–2112. [Google Scholar] [CrossRef]
- Li, M.; Zhu, D.; Wang, T.; Xia, X.; Tian, J.; Wang, S. Roles of Myeloid-Derived Suppressor Cell Subpopulations in Autoimmune Arthritis. Front. Immunol. 2018, 9, 2849. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Cane, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Luan, Y.; Mosheir, E.; Menon, M.C.; Wilson, D.; Woytovich, C.; Ochando, J.; Murphy, B. Monocytic myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4(+) Foxp3(+) Treg expansion. Am. J. Transplant. 2013, 13, 3123–3131. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, E.D.; Abrams, S.I. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Front. Immunol. 2020, 11, 1963. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Eruslanov, E.; Kubler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P.; et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: Link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immler, R.; Nadolni, W.; Bertsch, A.; Morikis, V.; Rohwedder, I.; Masgrau-Alsina, S.; Schroll, T.; Yevtushenko, A.; Soehnlein, O.; Moser, M.; et al. The voltage-gated potassium channel KV1.3 regulates neutrophil recruitment during inflammation. Cardiovasc. Res. 2022, 118, 1289–1302. [Google Scholar] [CrossRef] [PubMed]
- Cherny, V.V.; Markin, V.S.; DeCoursey, T.E. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J. Gen. Physiol. 1995, 105, 861–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanabe, A.; Okamura, Y. Effects of unsaturated fatty acids on the kinetics of voltage-gated proton channels heterologously expressed in cultured cells. J. Physiol. 2016, 594, 595–610. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Mokrab, Y.; Carvacho, I.; Sands, Z.A.; Sansom, M.S.P.; Clapham, D.E. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat. Struct. Mol. Biol. 2010, 17, 869–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, I.S.; Ruchti, E.; Kaczmarek, J.S.; Clapham, D.E. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. USA 2009, 106, 7642–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Gao, X.; Li, Y.; DeCoursey, T.E.; Shull, G.E.; Wang, H.S. The HVCN1 voltage-gated proton channel contributes to pH regulation in canine ventricular myocytes. J. Physiol. 2022, 600, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Wu, G.; Akhavan Sharif, M.R.; Baker, A.; Jia, Y.; Fahey, F.H.; Luo, H.R.; Feener, E.P.; Clapham, D.E. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 2012, 15, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Gattas, M.V.; Jaffe, A.; Barahona, J.; Conner, G.E. Proton channel blockers inhibit Duox activity independent of Hv1 effects. Redox Biol. 2020, 28, 101346. [Google Scholar] [CrossRef]
- Hondares, E.; Brown, M.A.; Musset, B.; Morgan, D.; Cherny, V.V.; Taubert, C.; Bhamrah, M.K.; Coe, D.; Marelli-Berg, F.; Gribben, J.G.; et al. Enhanced activation of an amino-terminally truncated isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18078–18083. [Google Scholar] [CrossRef] [Green Version]
- El Chemaly, A.; Okochi, Y.; Sasaki, M.; Arnaudeau, S.; Okamura, Y.; Demaurex, N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 2010, 207, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Petho, Z.; Balajthy, A.; Bartok, A.; Bene, K.; Somodi, S.; Szilagyi, O.; Rajnavolgyi, E.; Panyi, G.; Varga, Z. The anti-proliferative effect of cation channel blockers in T lymphocytes depends on the strength of mitogenic stimulation. Immunol. Lett. 2016, 171, 60–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Antibody | Clone | Company | Cat. No. | Application |
|---|---|---|---|---|---|
| 1 | Rat anti mouse CD16/CD32 | 2.4G2 | BD Pharmingen | 553141 | FC, FACS |
| 2 | Rat anti mouse CD11b-PE-Cy7 | M1/70 | eBioscience | 25-0112-82 | FACS |
| 3 | Rat anti CD11b-Alexa 594 | M1/70 | Biolegend | BZ 101254 | IF |
| 4 | Rat anti CD11b-PercP.Cy5.5 | M1/70 | eBiosciece | 101228 | FC |
| 5 | Rat anti Ly6C-APC | HK1.1 | Biolegend | 128015 | FACS, IF |
| 6 | Rat anti Ly6G-PE | 1A8 | BioLegend | 127608 | FACS |
| 7 | Rat anti Ly6G-APC | 1A8 | BioLegend | IF | |
| 8 | Rat anti MHCII-PercP-Cy5 | M5/114.15.2 | BioLegend | 107626 | FACS |
| 9 | Rat anti F4/80 -AF488 | CI:A3-1 | BioRad | MCA497A488 | FACS |
| 10 | Hamster anti mouse CD3e | 145-2C11 | Kind gift from Prof. Jo A. Van Ginderachter | T cell proliferation | |
| 11 | Rat anti mouse CD4-PE | RM4-5 | BD Pharmingen | 553049 | FC |
| 12 | Rat anti mouse CD8a-APC | clone53.6.7 | eBioscience | 17-0081-83 | FC |
| 13 | Rabbit anti mouse Hv1 | - | Alomone Labs | AHC-001 | WB, IF |
| 14 | Mouse anti β-actin | C4 | Santa Cruz Biotechnology | Sc-47778 | WB |
| 15 | Sheep anti mouse IgG-HRP linked | GE Healthcare | NA931 | WB | |
| 16 | Donkey anti rabbit IgG (H&L) Alexa488 preadsorbed | - | Abcam | ab150061 | IF |
| 17 | Donkey anti rabbit IgG-HRP linked | GE Healthcare | NA934 | WB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozzolino, M.; Gyöngyösi, A.; Korpos, E.; Gogolak, P.; Naseem, M.U.; Kállai, J.; Lanyi, A.; Panyi, G. The Voltage-Gated Hv1 H+ Channel Is Expressed in Tumor-Infiltrating Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2023, 24, 6216. https://doi.org/10.3390/ijms24076216
Cozzolino M, Gyöngyösi A, Korpos E, Gogolak P, Naseem MU, Kállai J, Lanyi A, Panyi G. The Voltage-Gated Hv1 H+ Channel Is Expressed in Tumor-Infiltrating Myeloid-Derived Suppressor Cells. International Journal of Molecular Sciences. 2023; 24(7):6216. https://doi.org/10.3390/ijms24076216
Chicago/Turabian StyleCozzolino, Marco, Adrienn Gyöngyösi, Eva Korpos, Peter Gogolak, Muhammad Umair Naseem, Judit Kállai, Arpad Lanyi, and Gyorgy Panyi. 2023. "The Voltage-Gated Hv1 H+ Channel Is Expressed in Tumor-Infiltrating Myeloid-Derived Suppressor Cells" International Journal of Molecular Sciences 24, no. 7: 6216. https://doi.org/10.3390/ijms24076216