
A Role of Sodium-Glucose Co-Transporter 2 in Cardiorenal Anemia Iron Deficiency Syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
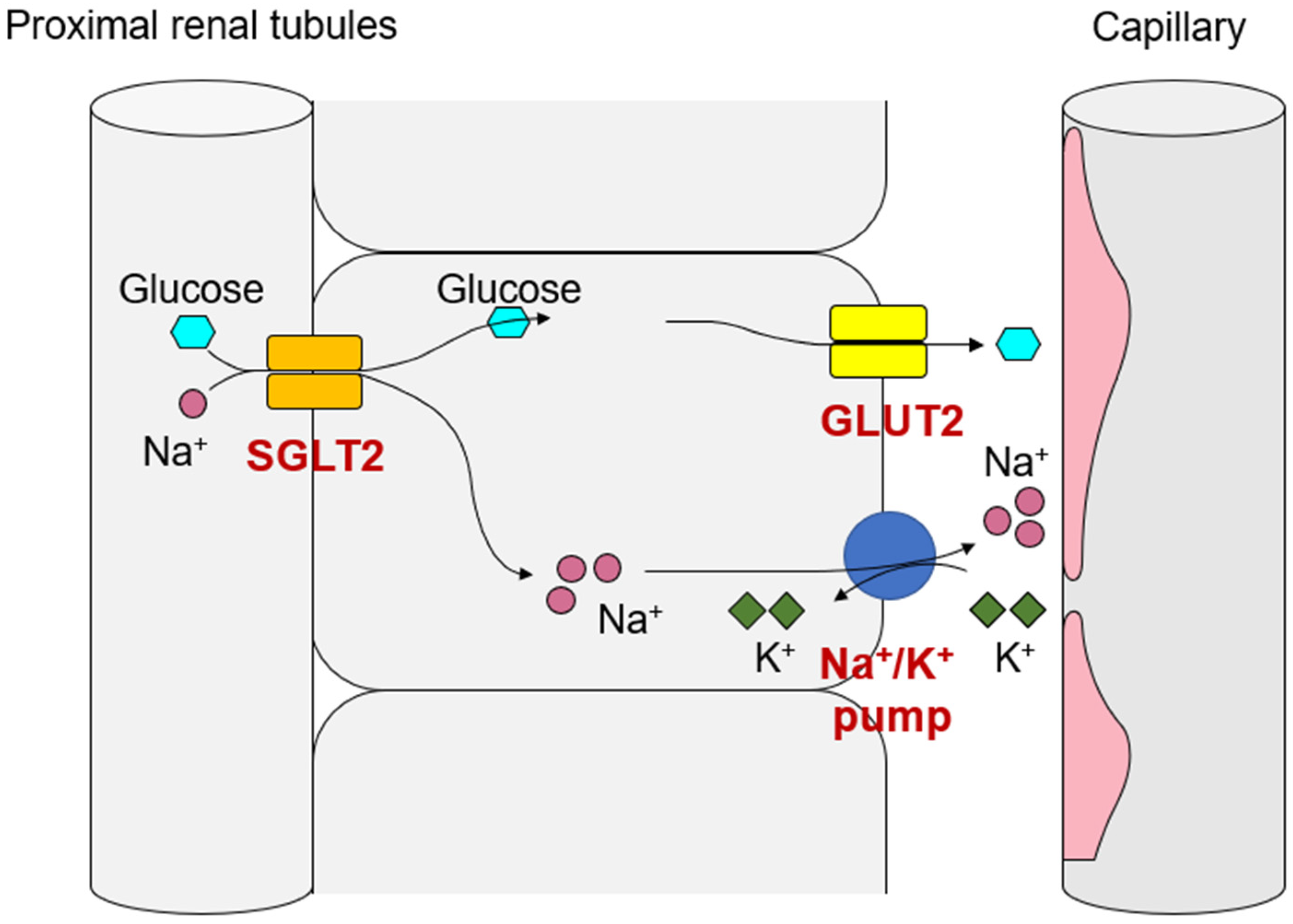
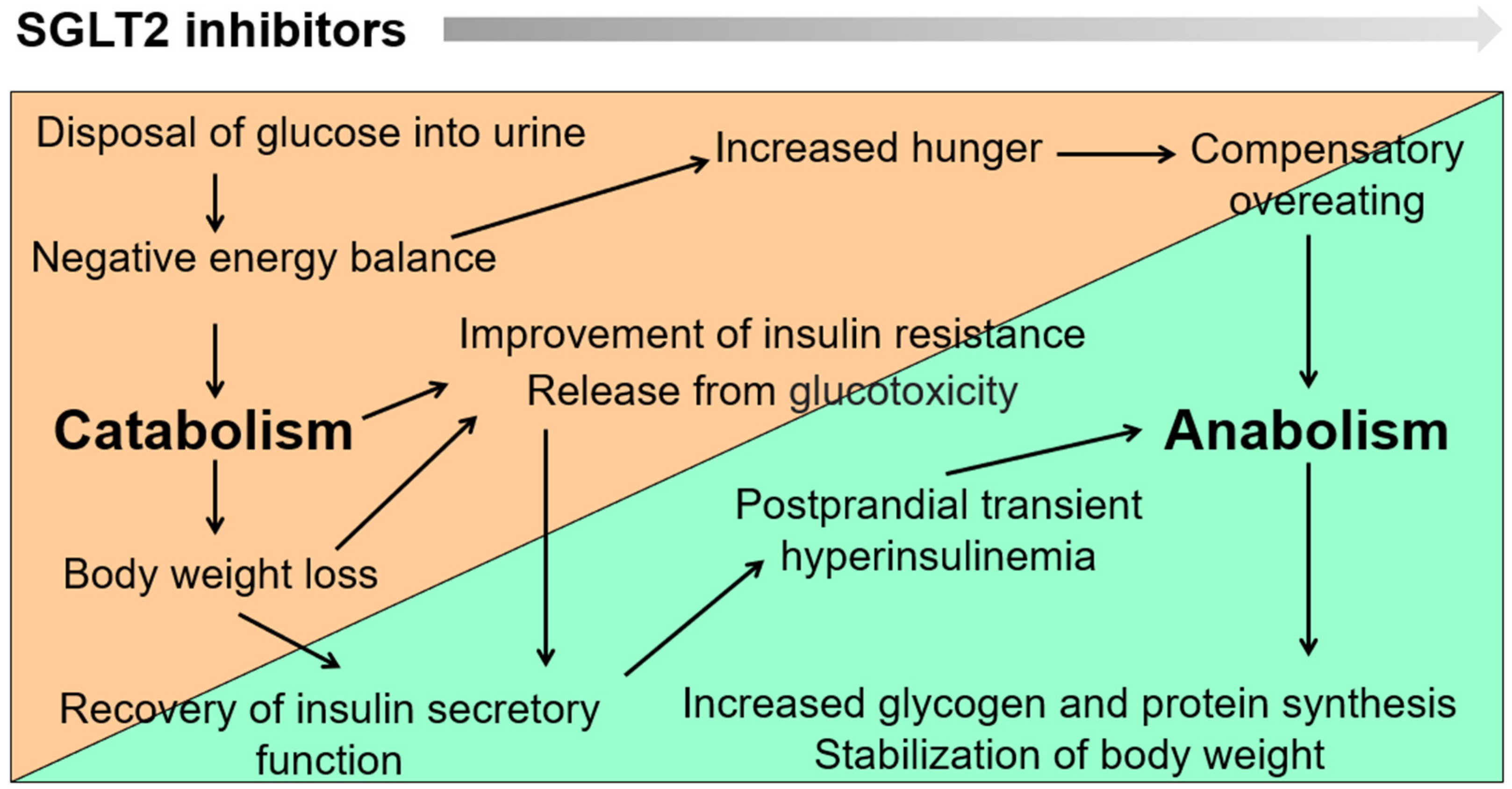
2. Dynamic Changes in Metabolic Set Points and Establishment of New Homeostasis by SGLT2 Blockade
3. Clinical Evidence for Cardiorenal Protection by SGLT2 Inhibition
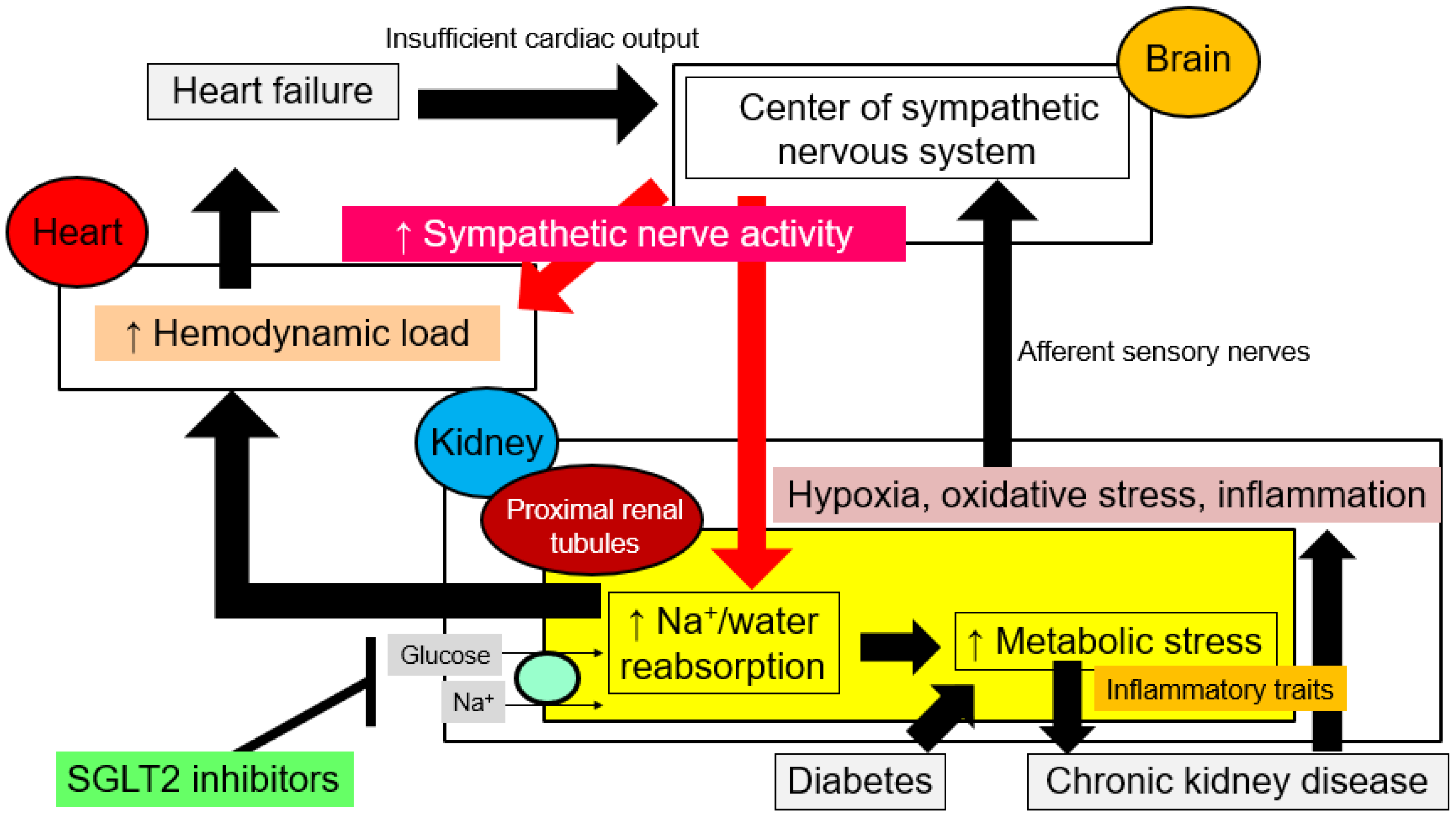
4. Correction by SGLT2 Inhibition of Aberrant Hemodynamics in Cardiorenal Syndrome
5. Iron Deficiency in Anemia
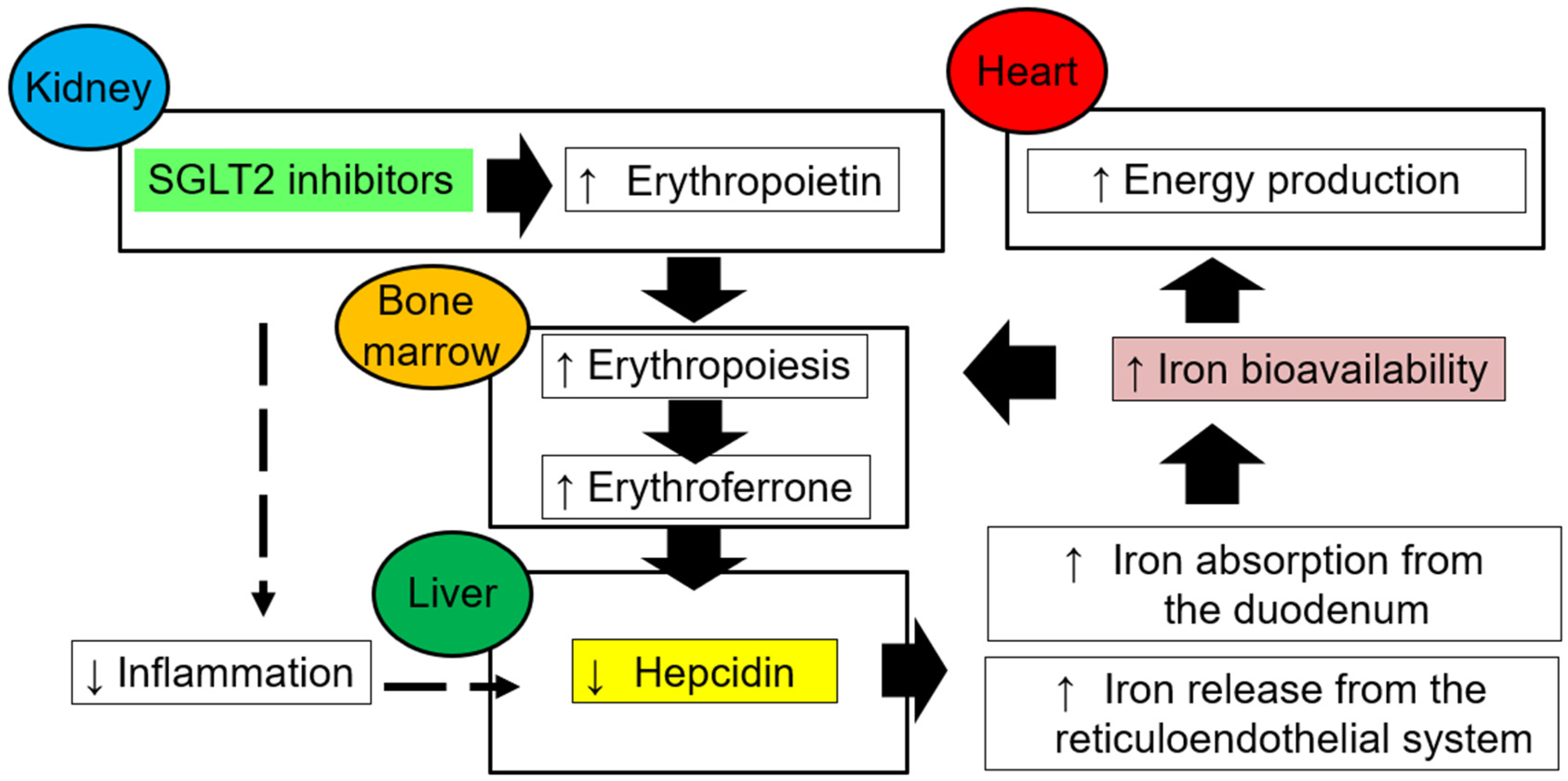
6. Restoration of Iron Bioavailability and Erythropoiesis with SGLT2 Blockade
7. Chronic Low-Level Inflammation in Cardiovascular Disease and CKD
8. Anti-Inflammatory Effect of SGLT2 Inhibitors
9. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sano, M.; Meguro, S.; Kawai, T.; Suzuki, Y. Increased grip strength with sodium-glucose cotransporter 2. J. Diabetes 2016, 8, 736–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Kosiborod, M.; Fitchett, D.; Wanner, C.; Hehnke, U.; Kaspers, S.; George, J.T.; Zinman, B. Improvement in Cardiovascular Outcomes with Empagliflozin Is Independent of Glycemic Control. Circulation 2018, 138, 1904–1907. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights from a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitchett, D.; Inzucchi, S.E.; Zinman, B.; Wanner, C.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Ofstad, A.P.; Salsali, A.; George, J.T.; et al. Mediators of the improvement in heart failure outcomes with empagliflozin in the EMPA-REG OUTCOME trial. ESC Heart Fail. 2021, 8, 4517–4527. [Google Scholar] [CrossRef] [PubMed]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraz-Muñoz, A.Y.; Weinstein, J.; Wald, R. eGFR Decline after SGLT2 Inhibitor Initiation: The Tortoise and the Hare Reimagined. Kidney360 2021, 2, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar]
- Mullens, W.; Verbrugge, F.H.; Nijst, P. Tang WHW. Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur. Heart J. 2017, 38, 1872–1882. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W.T. Hormones and hemodynamics in heart failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Sano, M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. Int. J. Mol. Sci. 2020, 21, 7676. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. Inter-organ Communication Pathway Manifested by Non-physiological Stress to the Kidney in Type II Diabetic Patients-Why Are Diabetic Patients Prone to Develop Heart Failure? Intern. Med. 2020, 59, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Sano, M. A Paradigm Shift in the Treatment of Type 2 Diabetes and Heart Failure. J. Atheroscler. Thromb. 2020, 27, 727–731. [Google Scholar] [CrossRef]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; Kiuchi, M.G.; Head, G.A.; Schlaich, M.P.; et al. SGLT2 Inhibitor-Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC Basic. Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef]
- Silverberg, D.S.; Wexler, D.; Blum, M.; Keren, G.; Sheps, D.; Leibovitch, E.; Brosh, D.; Laniado, S.; Schwartz, D.; Yachnin, T.; et al. The use of subcutaneous erythropoietin and intravenous iron for the treatment of the anemia of severe, resistant congestive heart failure improves cardiac and renal function and functional cardiac class, and markedly reduces hospitalizations. J. Am. Coll. Cardiol. 2000, 35, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
- Swedberg, K.; McMurray, J.J.; Young, J.B. Darbepoetin alfa in systolic heart failure. N. Engl. J. Med. 2013, 369, 488–489. [Google Scholar]
- Okonko, D.O.; Mandal, A.K.; Missouris, C.G.; Poole-Wilson, P.A. Disordered iron homeostasis in chronic heart failure: Prevalence, predictors, and relation to anemia, exercise capacity, and survival. J. Am. Coll. Cardiol. 2011, 58, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Kirwan, B.A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: A multicentre, double-blind, randomised, controlled trial. Lancet 2020, 396, 1895–1904. [Google Scholar] [CrossRef]
- Lewis, G.D.; Malhotra, R.; Hernandez, A.F.; McNulty, S.E.; Smith, A.; Felker, G.M.; Tang, W.H.W.; LaRue, S.J.; Redfield, M.M.; Semigran, M.J.; et al. Effect of Oral Iron Repletion on Exercise Capacity in Patients with Heart Failure with Reduced Ejection Fraction and Iron Deficiency: The IRONOUT HF Randomized Clinical Trial. JAMA 2017, 317, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jamieson, K.L.; Grenier, J.; Nikhanj, A.; Tang, Z.; Wang, F.; Wang, S.; Seidman, J.G.; Seidman, C.E.; Thompson, R.; et al. Myocardial Iron Deficiency and Mitochondrial Dysfunction in Advanced Heart Failure in Humans. J. Am. Heart Assoc. 2022, 11, e022853. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Anker, S.D.; Butler, J.; Filippatos, G.; Iwata, T.; Salsali, A.; Zeller, C.; Pocock, S.J.; Zannad, F.; Packer, M. Impact of anaemia and the effect of empagliflozin in heart failure with reduced ejection fraction: Findings from EMPEROR-Reduced. Eur. J. Heart Fail. 2022, 24, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Neuen, B.L.; Jardine, M.J.; Bakris, G.; Edwards, R.; Levin, A.; Mahaffey, K.W.; Neal, B.; Pollock, C.; Rosenthal, N.; et al. Effects of canagliflozin on anaemia in patients with type 2 diabetes and chronic kidney disease: A post-hoc analysis from the CREDENCE trial. Lancet Diabetes Endocrinol. 2020, 8, 903–914. [Google Scholar] [CrossRef]
- Vlahakos, V.; Marathias, K.; Lionaki, S.; Loukides, S.; Zakynthinos, S.; Vlahakos, D. The paradigm shift from polycythemia to anemia in COPD: The critical role of the renin-angiotensin system inhibitors. Expert Rev. Respir. Med. 2022, 16, 391–398. [Google Scholar] [CrossRef]
- Murashima, M.; Tanaka, T.; Kasugai, T.; Tomonari, T.; Ide, A.; Ono, M.; Mizuno, M.; Suzuki, T.; Hamano, T. Sodium-glucose cotransporter 2 inhibitors and anemia among diabetes patients in real clinical practice. J. Diabetes Investig. 2022, 13, 638–646. [Google Scholar] [CrossRef]
- Docherty, K.F.; Welsh, P.; Verma, S.; De Boer, R.A.; O’Meara, E.; Bengtsson, O.; Køber, L.; Kosiborod, M.N.; Hammarstedt, A.; Langkilde, A.M.; et al. Iron Deficiency in Heart Failure and Effect of Dapagliflozin: Findings from DAPA-HF. Circulation 2022, 146, 980–994. [Google Scholar] [CrossRef]
- Mazer, C.D.; Hare, G.M.T.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Jüni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores, and Red Blood Cell Morphology in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2020, 141, 704–707. [Google Scholar] [CrossRef]
- Packer, M. Alleviation of functional iron deficiency by SGLT2 inhibition in patients with type 2 diabetes. Diabetes Obes. Metab. 2022. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. How can sodium-glucose cotransporter 2 inhibitors stimulate erythrocytosis in patients who are iron-deficient? Implications for understanding iron homeostasis in heart failure. Eur. J. Heart Fail. 2022, 24, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Szklarz, M.; Gontarz-Nowak, K.; Matuszewski, W.; Bandurska-Stankiewicz, E. Can Iron Play a Crucial Role in Maintaining Cardiovascular Health in the 21st Century? Int. J. Environ. Res. Public Health 2022, 19, 11990. [Google Scholar] [CrossRef]
- Silverberg, D.S.; Wexler, D.; Blum, M.; Iaina, A. The cardio renal anemia syndrome: Correcting anemia in patients with resistant congestive heart failure can improve both cardiac and renal function and reduce hospitalizations. Clin. Nephrol. 2003, 60 (Suppl. S1), S93–S102. [Google Scholar] [PubMed]
- Cappellini, M.D.; Comin-Colet, J.; de Francisco, A.; Dignass, A.; Doehner, W.; Lam, C.S.; Macdougall, I.C.; Rogler, G.; Camaschella, C.; Kadir, R.; et al. Iron deficiency across chronic inflammatory conditions: International expert opinion on definition, diagnosis and management. Am. J. Hematol. 2017, 92, 1068–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, C.; Carbonara, R.; Ruggieri, R.; Passantino, A.; Scrutinio, D. Iron Deficiency: A New Target for Patients with Heart Failure. Front. Cardiovasc. Med. 2021, 8, 709872. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Packer, M. Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure. J. Am. Coll. Cardiol. Heart Fail. 2023, 11, 106–114. [Google Scholar] [CrossRef]
- Masini, G.; Graham, F.J.; Pellicori, P.; Cleland, J.G.F.; Cuthbert, J.J.; Kazmi, S.; Inciardi, R.M.; Clark, A.L. Criteria for Iron Deficiency in Patients with Heart Failure. J. Am. Coll. Cardiol. 2022, 79, 341–351. [Google Scholar] [CrossRef]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Souma, T.; Yamazaki, S.; Moriguchi, T.; Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Abe, M.; Kiyomoto, H.; Ito, S.; et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1599–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Everett, B.M. Novel Antiatherosclerotic Therapies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.I.; Masood, S.; Furlanetto, D.M.; Nicolaou, S. Urate Crystals; Beyond Joints. Front. Med. 2021, 8, 649505. [Google Scholar] [CrossRef]
- Shirakawa, K.; Sano, M. Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: An Overview and Potential Therapeutic Approaches. Biomedicines 2022, 10, 1850. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Tinti, F.; Lai, S.; Noce, A.; Rotondi, S.; Marrone, G.; Mazzaferro, S.; Di Daniele, N.; Mitterhofer, A.P. Chronic Kidney Disease as a Systemic Inflammatory Syndrome: Update on Mechanisms Involved and Potential Treatment. Life 2021, 11, 419. [Google Scholar] [PubMed]
- Wolkow, P.P.; Niewczas, M.A.; Perkins, B.; Ficociello, L.H.; Lipinski, B.; Warram, J.H.; Krolewski, A.S. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J. Am. Soc. Nephrol. 2008, 19, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From RESCUE to ZEUS: Will interleukin-6 inhibition with ziltivekimab. Cardiovasc. Res. 2021, 117, e138–e140. [Google Scholar] [CrossRef]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic repro-gramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Sano, M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells 2021, 10, 2435. [Google Scholar] [CrossRef]
- Shirakawa, K.; Sano, M. Drastic transformation of visceral adipose tissue and peripheral CD4 T cells in obesity. Front. Immunol. 2023, 13, 1044737. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, T.E.; Kovanen, P.T. Coronary artery disease: ‘gout’ in the artery? Eur. Heart J. 2021, 42, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanashi, T.; Iwata, M.; Kamiya, N.; Tsunetomi, K.; Kajitani, N.; Wada, N.; Iitsuka, T.; Yamauchi, T.; Miura, A.; Pu, S.; et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 2017, 7, 7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodbard, H.W.; Rosenstock, J.; Canani, L.H.; Deerochanawong, C.; Gumprecht, J.; Lindberg, S.Ø.; Lingvay, I.; Søndergaard, A.L.; Treppendahl, M.B.; Montanya, E. Oral Semaglutide versus Empagliflozin in Patients with Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care 2019, 42, 2272–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano, M. A Role of Sodium-Glucose Co-Transporter 2 in Cardiorenal Anemia Iron Deficiency Syndrome. Int. J. Mol. Sci. 2023, 24, 5983. https://doi.org/10.3390/ijms24065983
Sano M. A Role of Sodium-Glucose Co-Transporter 2 in Cardiorenal Anemia Iron Deficiency Syndrome. International Journal of Molecular Sciences. 2023; 24(6):5983. https://doi.org/10.3390/ijms24065983
Chicago/Turabian StyleSano, Motoaki. 2023. "A Role of Sodium-Glucose Co-Transporter 2 in Cardiorenal Anemia Iron Deficiency Syndrome" International Journal of Molecular Sciences 24, no. 6: 5983. https://doi.org/10.3390/ijms24065983