

Ultrastructural Evidence of Mitochondrial Dysfunction in Osteomyelitis Patients
 , , and
, , and
Abstract
:1. Introduction
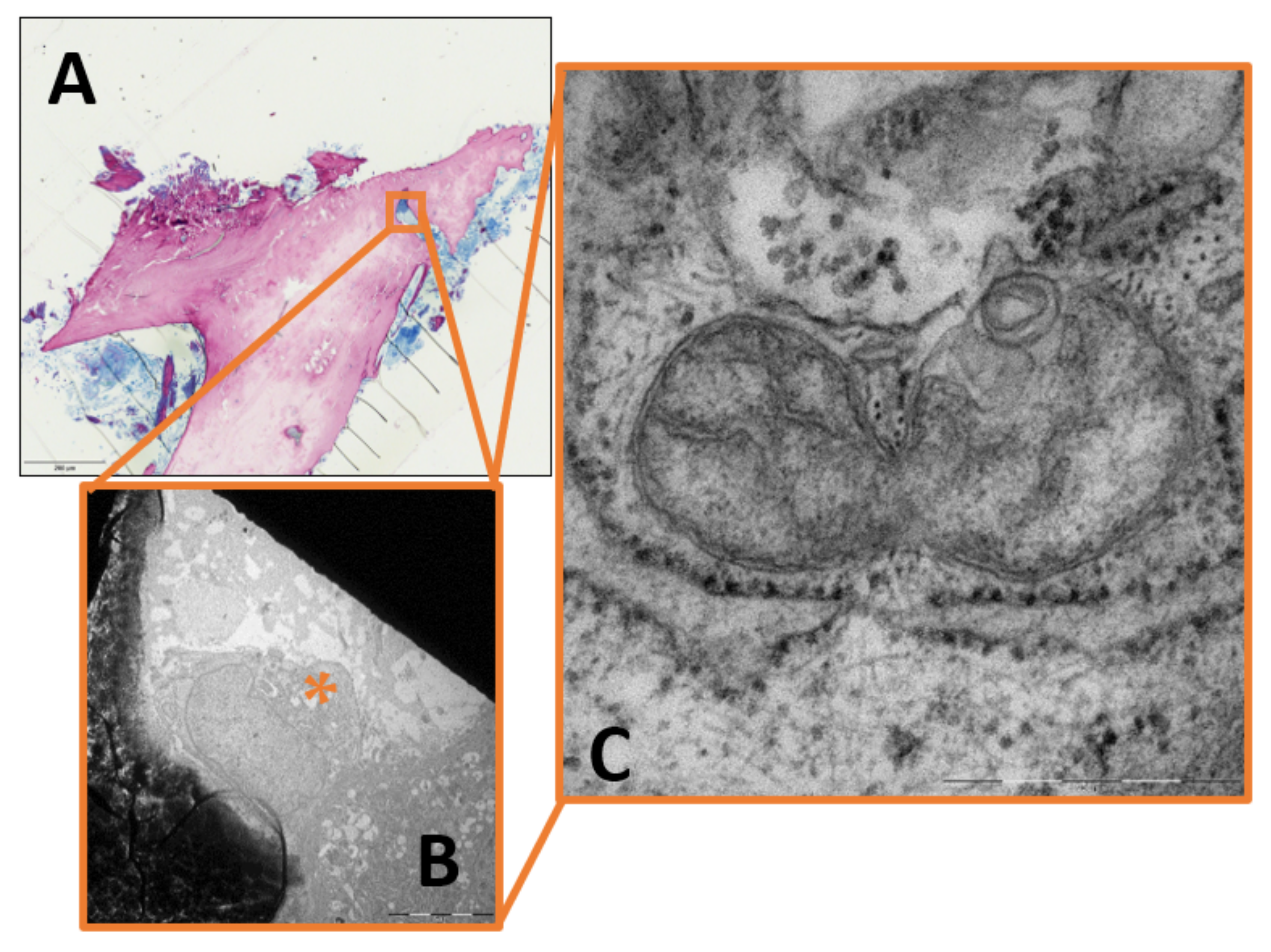
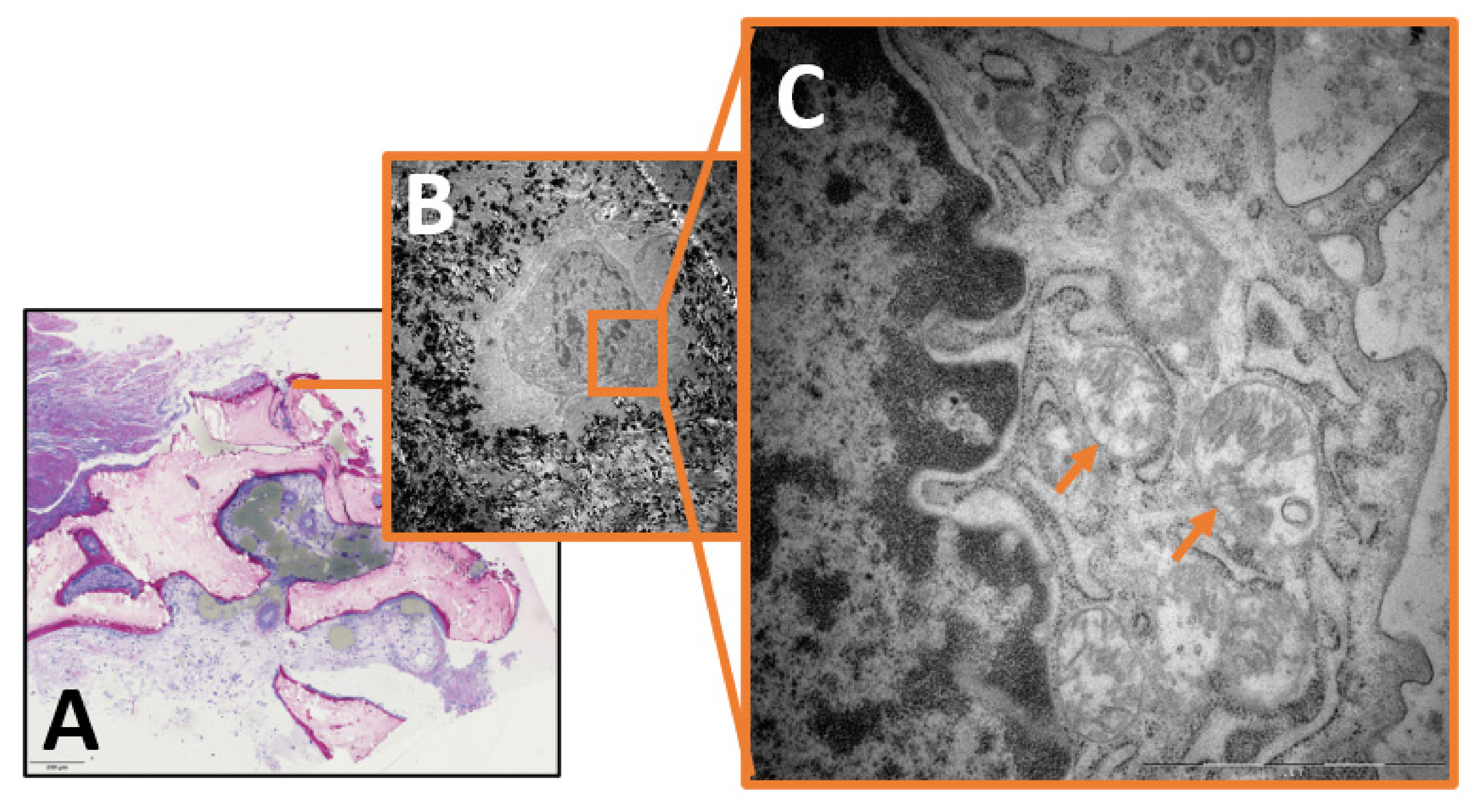
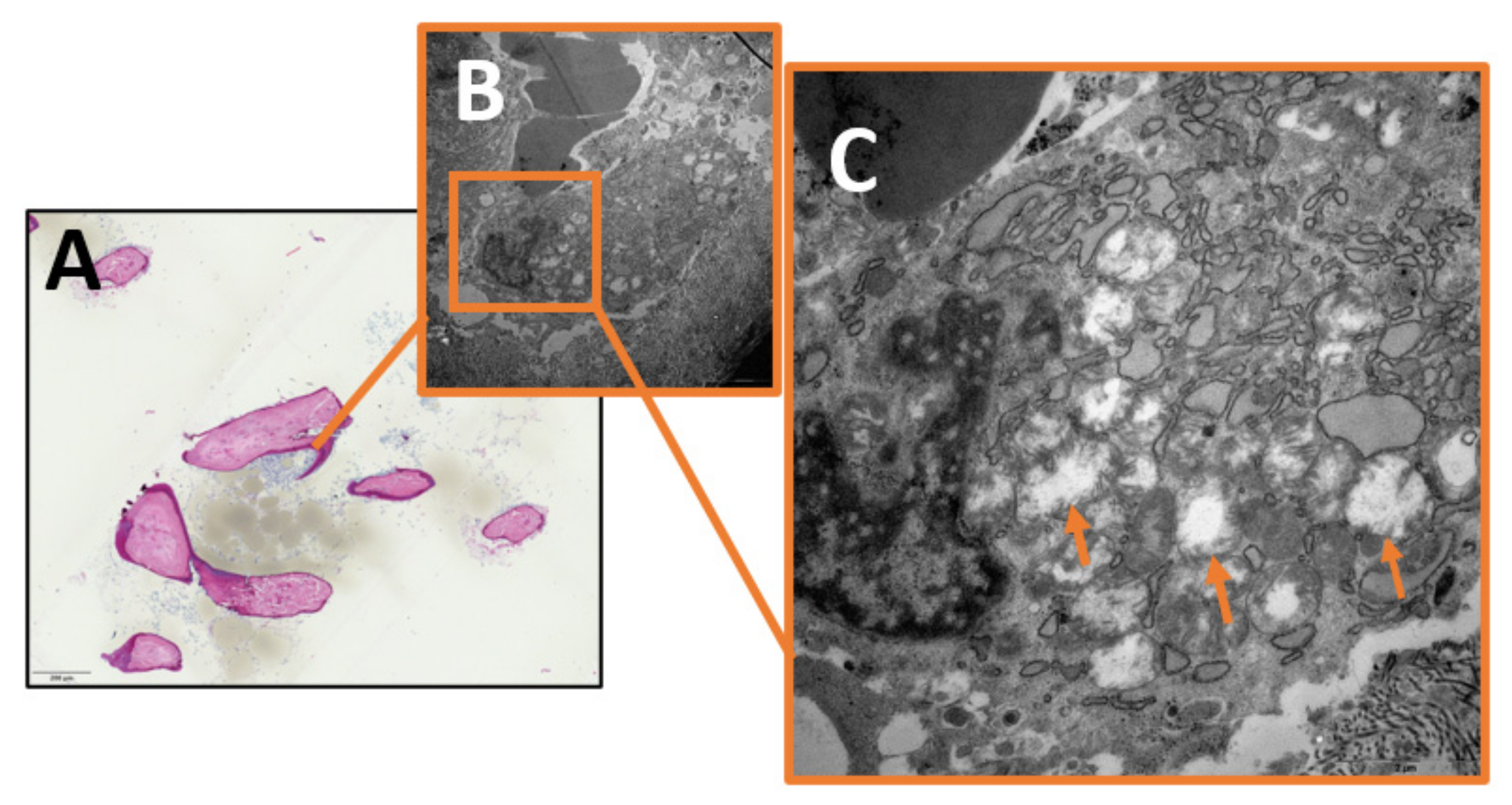
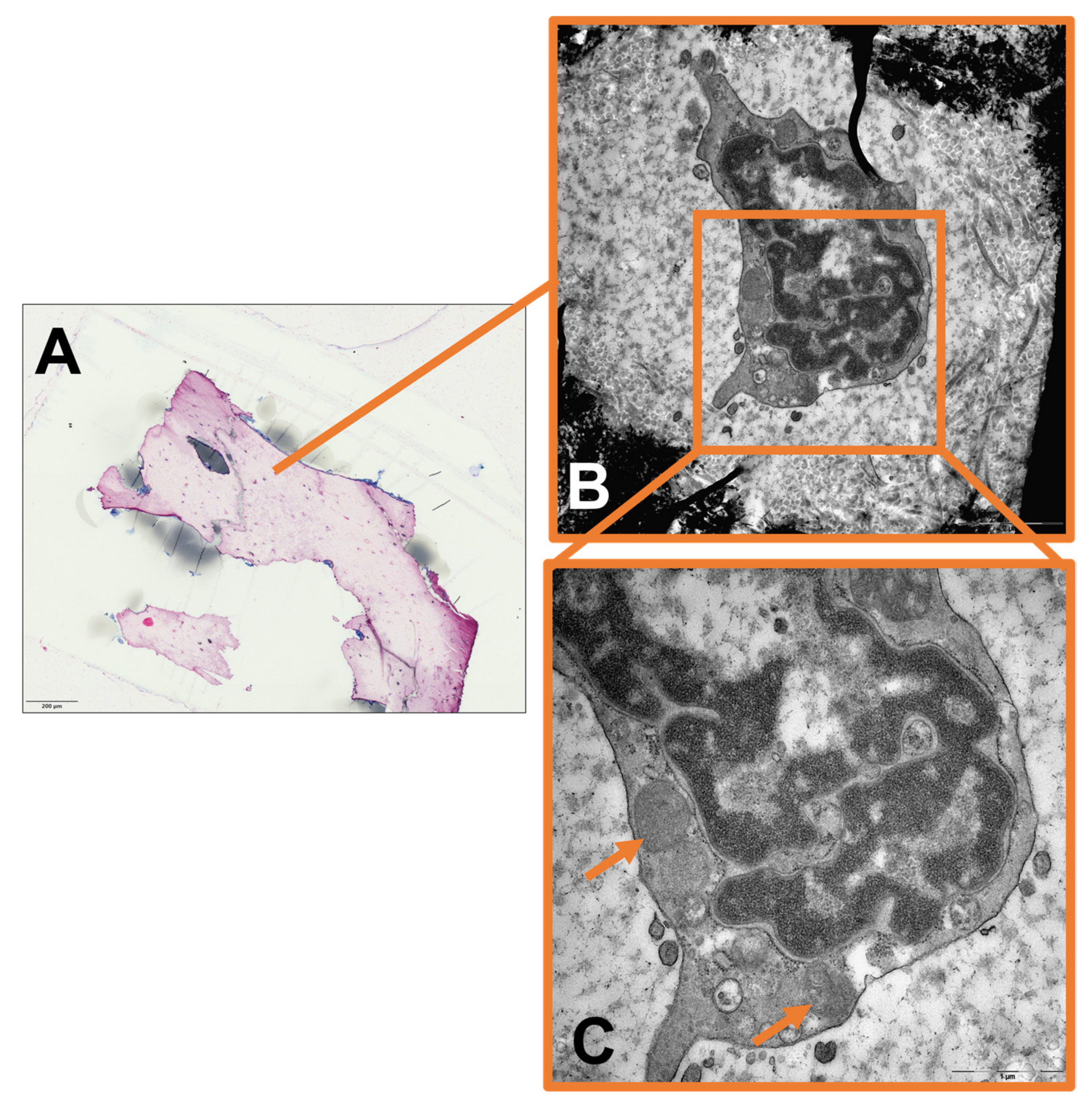
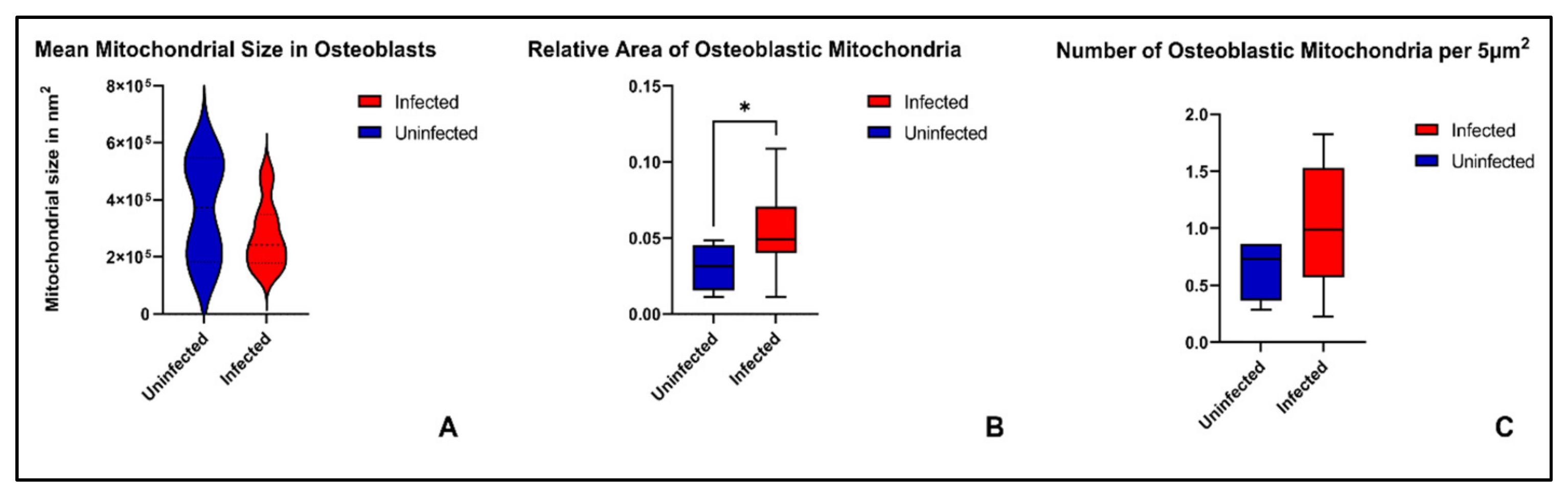
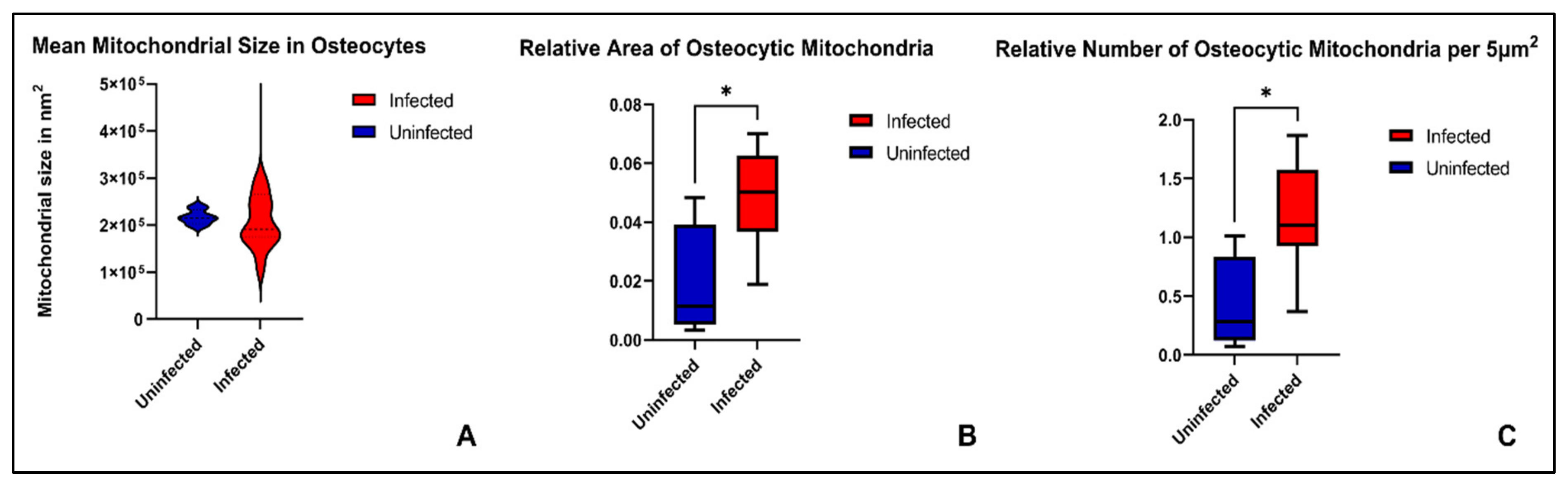
2. Results
3. Discussion
3.1. Mitochondrial Dysfunction and Oxidative Stress in Chronic Osteomyelitis
3.2. Mitochondrial Fragmentation in Chronic Osteomyelitis
3.3. The Role of Intracellular Staph. aureus in Chronic Osteomyelitis
3.4. Limitations
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ennker, I.C.; Ennker, J.C. The history of the management of sternal osteomyelitis and mediastinitis—From Hippocrates until today. GMS Interdiscip. Plast. Reconstr. Surg. DGPW 2014, 3, Doc07. [Google Scholar] [CrossRef]
- Rupp, M.; Alt, V.; Lowenberg, D. Management of Acute and Chronic Osteomyelitis. In Orthopaedic Knowledge Update: Trauma 6, 6th ed.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2022; pp. 149–167. ISBN 978-1-975163-68-6. [Google Scholar]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimza, B.D.; Cassat, J.E. Mechanisms of Antibiotic Failure During Staphylococcus aureus Osteomyelitis. Front. Immunol. 2021, 12, 638085. [Google Scholar] [CrossRef] [PubMed]
- Zelmer, A.R.; Nelson, R.; Richter, K.; Atkins, G.J. Can intracellular Staphylococcus aureus in osteomyelitis be treated using current antibiotics? A systematic review and narrative synthesis. Bone Res. 2022, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.; Mendelsohn, D.; Brochhausen, C.; Rupp, M.; Alt, V. Intracellular S. aureus in Osteoblasts in a Clinical Sample from a Patient with Chronic Osteomyelitis—A Case Report. Pathogens 2021, 10, 1064. [Google Scholar] [CrossRef] [PubMed]
- Ferver, A.; Greene, E.; Wideman, R.; Dridi, S. Evidence of Mitochondrial Dysfunction in Bacterial Chondronecrosis With Osteomyelitis–Affected Broilers. Front. Veter.-Sci. 2021, 8, 640901. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, D.H.; Schnabel, K.; Mamilos, A.; Sossalla, S.; Pabel, S.; Duerr, G.D.; Keller, K.; Schmitt, V.H.; Barsch, F.; Walter, N.; et al. Structural Analysis of Mitochondrial Dynamics—From Cardiomyocytes to Osteoblasts: A Critical Review. Int. J. Mol. Sci. 2022, 23, 4571. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.; Neuman, N. The Mighty Mitochondria. Mol. Cell 2016, 61, 641. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; Hart, L.M.; van Essen, E.; Heine, R.J.; Nijpels, G.; Tafrechi, R.S.J.; Raap, A.K.; Janssen, G.M.; Lemkes, H.H. Mitochondrial Diabetes: Molecular mechanisms and clinical presentation. Diabetes 2004, 53 (Suppl 1), S103–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Genestier, A.-L.; Michallet, M.-C.; Prévost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.; et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lin, Y.; Zong, Y.; Ma, X.; Jiang, C.; Shan, H.; Xia, W.; Yin, L.; Wang, N.; Zhou, L.; et al. Staphylococcus aureus Infection Initiates Hypoxia-Mediated Transforming Growth Factor-β1 Upregulation to Trigger Osteomyelitis. Msystems 2022, 7, e0038022. [Google Scholar] [CrossRef] [PubMed]
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Zhang, X.; Ding, T.; Zhong, Z.; Hu, H.; Xu, Z.; Deng, J. Mitochondrial Dynamics Imbalance: A Strategy for Promoting Viral Infection. Front. Microbiol. 2020, 11, 1992. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S.; Paz, M.V.; Cotán, D.; Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; de La Mata, M.; Pavón, A.D.; de Lavera, I.; et al. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.-B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Ding, C. Mitochondria in Cryptococcus: An update of mitochondrial transcriptional regulation in Cryptococcus. Curr. Genet. 2023, 69, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [Green Version]
- Charlson, M.E.; Wells, M.T.; Ullman, R.; King, F.; Shmukler, C. The Charlson Comorbidity Index Can Be Used Prospectively to Identify Patients Who Will Incur High Future Costs. PLoS ONE 2014, 9, e112479. [Google Scholar] [CrossRef] [Green Version]
- Lyamzaev, K.G.; Knorre, D.A.; Chernyak, B.V. Mitoptosis, Twenty Years After. Biochemistry 2020, 85, 1484–1498. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch. Biochem. Biophys. 2017, 630, 1–8. [Google Scholar] [CrossRef]
- Chang, X.; Liu, R.; Li, R.; Peng, Y.; Zhu, P.; Zhou, H. Molecular Mechanisms of Mitochondrial Quality Control in Ischemic Cardiomyopathy. Int. J. Biol. Sci. 2023, 19, 426–448. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Tandler, B.; Hoppel, C.L. Mitochondrial Division in Rat Cardiomyocytes: An Electron Microscope Study. Anat. Rec. 2012, 295, 1455–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devraj, G.; Beerlage, C.; Brüne, B.; Kempf, V.A. Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. 2017, 19, 144–156. [Google Scholar] [CrossRef]
- Schaffer, K.; Taylor, C.T. The impact of hypoxia on bacterial infection. FEBS J. 2015, 282, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Eckl, E.-M.; Ziegemann, O.; Krumwiede, L.; Fessler, E.; Jae, L.T. Sensing, signaling and surviving mitochondrial stress. Cell. Mol. Life Sci. 2021, 78, 5925–5951. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Takano, Y.; Satrialdi; Abe, J.; Hibino, M.; Harashima, H. Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress. Biomolecules 2020, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCully, J.D.; Cowan, D.B.; Emani, S.M.; del Nido, P.J. Mitochondrial transplantation: From animal models to clinical use in humans. Mitochondrion 2017, 34, 127–134. [Google Scholar] [CrossRef]
- Park, A.; Oh, M.; Lee, S.; Oh, K.-J.; Lee, E.-W.; Lee, S.; Bae, K.-H.; Han, B.; Kim, W. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guo, S.; Tang, Y.; Mou, C.; Hu, X.; Shao, F.; Yan, W.; Wu, Q. Mitochondrial Fusion and Fission in Neuronal Death Induced by Cerebral Ischemia-Reperfusion and Its Clinical Application: A Mini-Review. Med. Sci. Monit. 2020, 26, e928651. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yu, Y.; Luo, X.; Wang, J.; Lan, X.; Liu, P.; Feng, Y.; Jian, W. Myocardial Ischemia-Reperfusion Injury: Therapeutics from a Mitochondria-Centric Perspective. Cardiology 2021, 146, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free. Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [Green Version]
- Hart, B.B. Hyperbaric oxygen for refractory osteomyelitis. Undersea Hyperb. Med. J. Undersea Hyperb. Med. Soc. Inc. 2021, 48, 297–321. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.; Han, L.; Ambrogini, E.; Bartell, S.M.; Manolagas, S.C. Oxidative Stress Stimulates Apoptosis and Activates NF-κB in Osteoblastic Cells via a PKCβ/p66shc Signaling Cascade: Counter Regulation by Estrogens or Androgens. Mol. Endocrinol. 2010, 24, 2030–2037. [Google Scholar] [CrossRef] [Green Version]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Liò, P.; Paoletti, N.; Moni, M.A.; Atwell, K.; Merelli, E.; Viceconti, M. Modelling osteomyelitis. BMC Bioinform. 2012, 13, S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Gan, X.; He, Y.; Zhu, Z.; Zhu, J.; Yu, H. Drp1-dependent mitochondrial fission mediates osteogenic dysfunction in inflammation through elevated production of reactive oxygen species. PLoS ONE 2017, 12, e0175262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, V.; Possetti, V.; Parente, R.; Bottazzi, B.; Inforzato, A.; Sobacchi, C. The osteoblast secretome in Staphylococcus aureus osteomyelitis. Front. Immunol. 2022, 13, 1048505. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Furusho, H.; Rider, D.B.; Dobeck, J.M.; Kuo, W.P.; Fujimura, A.; Yoganathan, S.; Hirai, K.; Xu, S.; Sasaki, K.; et al. Endodontic Infection–induced Inflammation Resembling Osteomyelitis of the Jaws in Toll-like Receptor 2/Interleukin 10 Double-knockout Mice. J. Endod. 2019, 45, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Cooper, K.F.; Strich, R. The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy. Biology 2021, 10, 33. [Google Scholar] [CrossRef]
- Disatnik, M.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute Inhibition of Excessive Mitochondrial Fission After Myocardial Infarction Prevents Long-term Cardiac Dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Dong, Q.; Liu, Z.; Liu, Z.; Qu, Y.; Li, X.; Huo, C.; Jia, X.; Fu, F.; Wang, X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc. Diabetol. 2017, 16, 19. [Google Scholar] [CrossRef] [Green Version]
- Bosse, M.J.; Gruber, H.E.; Ramp, W.K. Internalization of Bacteria by Osteoblasts in a Patient with Recurrent, Long-Term Osteomyelitis: A Case Report. J. Bone Jt. Surg. 2005, 87, 1343–1347. [Google Scholar] [CrossRef]
- Nesson, E.T.; McDowell, S.A. Innovations in Evaluating Statin Benefit and Efficacy in Staphylococcus aureus Intracellular Infection Management. Int. J. Mol. Sci. 2022, 23, 13006. [Google Scholar] [CrossRef]
- Urish, K.L.; Cassat, J.E. Staphylococcus aureus Osteomyelitis: Bone, Bugs, and Surgery. Infect. Immun. 2020, 88, e00932-19. [Google Scholar] [CrossRef] [PubMed]
- Watkins, K.E.; Unnikrishnan, M. Chapter Three—Evasion of host defenses by intracellular Staphylococcus aureus. In Advances in Applied Microbiology; Gadd, G.M., Sariaslani, S., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 105–141. ISBN 0065-2164. [Google Scholar]
- Sybenga, A.B.; Jupiter, D.C.; Speights, V.; Rao, A. Diagnosing Osteomyelitis: A Histology Guide for Pathologists. J. Foot Ankle Surg. 2020, 59, 75–85. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.; Sousa, R.; Wouthuyzen-Bakker, M.; Chen, A.F.; Soriano, A.; Vogely, H.C.; Clauss, M.; Higuera, C.A.; Trebše, R. The EBJIS definition of periprosthetic joint infection. Bone Jt. J. 2021, 103-B, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Govaert, G.A.M.; Kuehl, R.; Atkins, B.L.; Trampuz, A.; Morgenstern, M.; Obremskey, W.T.; Verhofstad, M.H.J.; McNally, M.A.; Metsemakers, W.-J. Diagnosing Fracture-Related Infection: Current Concepts and Recommendations. J. Orthop. Trauma 2020, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Walter, N.; Baertl, S.; Lang, S.; Lowenberg, D.W.; Alt, V. Terminology of bone and joint infection. Bone Jt. Res. 2021, 10, 742–743. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E. The ultrastructure of the osteocyte. In Ultrastructure of Skeletal Tissues; Springer: Boston, MA, USA, 1990; pp. 223–237. [Google Scholar]
- Scherft, J.P.; Groot, C.G. The electron microscopic structure of the osteoblast. In Ultrastructure of Skeletal Tissues; Springer: Boston, MA, USA, 1990; pp. 209–222. [Google Scholar] [CrossRef]
- Huang, C.; Deng, K.; Wu, M. Mitochondrial cristae in health and disease. Int. J. Biol. Macromol. 2023, 235. [Google Scholar] [CrossRef]
- Tiemann, A.; Hofmann, G.O.; Krukemeyer, M.G.; Krenn, V.; Langwald, S. Histopathological Osteomyelitis Evaluation Score (HOES)—An innovative approach to histopathological diagnostics and scoring of osteomyelitis. GMS Interdiscip. Plast. Reconstr. Surg. DGPW 2014, 3, Doc08. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Osteoblasts Analyzed | Osteocytes Analyzed | Age | Gender | Entity | Location | CCI * | Most Recent Pathogen |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 | 6 | 62 | Male | OM | Tibia | 4 | MSSA |
| 2 | 1 | 6 | 50 | Female | OM | First metatarsal | 2 | MSSA; Serratia ureilytica |
| 3 | 4 | 8 | 83 | Male | PJI | Femur | 2 | MSSA |
| 4 | 3 | 16 | 63 | Male | FRI | Femur | 1 | Staph. haemolyticus; Staph. epidermidis; VRE |
| 5 | 8 | 7 | 72 | Male | PJI | Femur | 6 | Staph. epidermidis |
| 6 | 1 | 14 | 56 | Male | FRI | Calcaneus | 0 | MSSA; Bacillus cereus |
| 7 | 4 | 3 | 59 | Male | PJI | Tibia | 1 | MSSA; Morganella morganii; E. coli |
| 8 | 1 | 3 | 66 | Female | FRI | Tibia | 0 | Mycobacterium abscessus |
| 9 | 7 | 11 | 57 | Male | FRI | Tibia | 2 | Staph. epidermidis |
| 10 | 14 | 3 | 17 | Male | OM | Femur | 0 | MSSA |
| 11 | 8 | 3 | 40 | Male | FRI | Femur | 0 | MSSA; Stenotrophomonas maltophilia |
| 12 | 4 | 4 | 71 | Male | PJI | Femur | 1 | Staph. epidermidis; Entero-coccus faecium |
| 13 | 0 | 9 | 14 | Male | OM | Tibia | 1 | MRSA |
| 14 | 3 | 0 | 71 | Male | PJI | Femur | 5 | Streptococcus agalactiae |
| Control | ||||||||
| 1 | 7 | 15 | 68 | Female | - | Femur | 1 | - |
| 2 | 5 | 2 | 53 | Male | - | Tibia | 0 | - |
| 3 | 2 | 11 | 67 | Male | - | Pelvis | 0 | - |
| 4 | 2 | 6 | 41 | Male | - | Femur | 0 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendelsohn, D.H.; Niedermair, T.; Walter, N.; Alt, V.; Rupp, M.; Brochhausen, C. Ultrastructural Evidence of Mitochondrial Dysfunction in Osteomyelitis Patients. Int. J. Mol. Sci. 2023, 24, 5709. https://doi.org/10.3390/ijms24065709
Mendelsohn DH, Niedermair T, Walter N, Alt V, Rupp M, Brochhausen C. Ultrastructural Evidence of Mitochondrial Dysfunction in Osteomyelitis Patients. International Journal of Molecular Sciences. 2023; 24(6):5709. https://doi.org/10.3390/ijms24065709
Chicago/Turabian StyleMendelsohn, Daniel H., Tanja Niedermair, Nike Walter, Volker Alt, Markus Rupp, and Christoph Brochhausen. 2023. "Ultrastructural Evidence of Mitochondrial Dysfunction in Osteomyelitis Patients" International Journal of Molecular Sciences 24, no. 6: 5709. https://doi.org/10.3390/ijms24065709