
The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges
 , and
, and
Abstract
:1. Introduction
2. Characterization of a Healthy Core Blood Microbiome
3. Clinical Significance of the Blood Microbiome in Human Diseases
3.1. Cardiometabolic Diseases

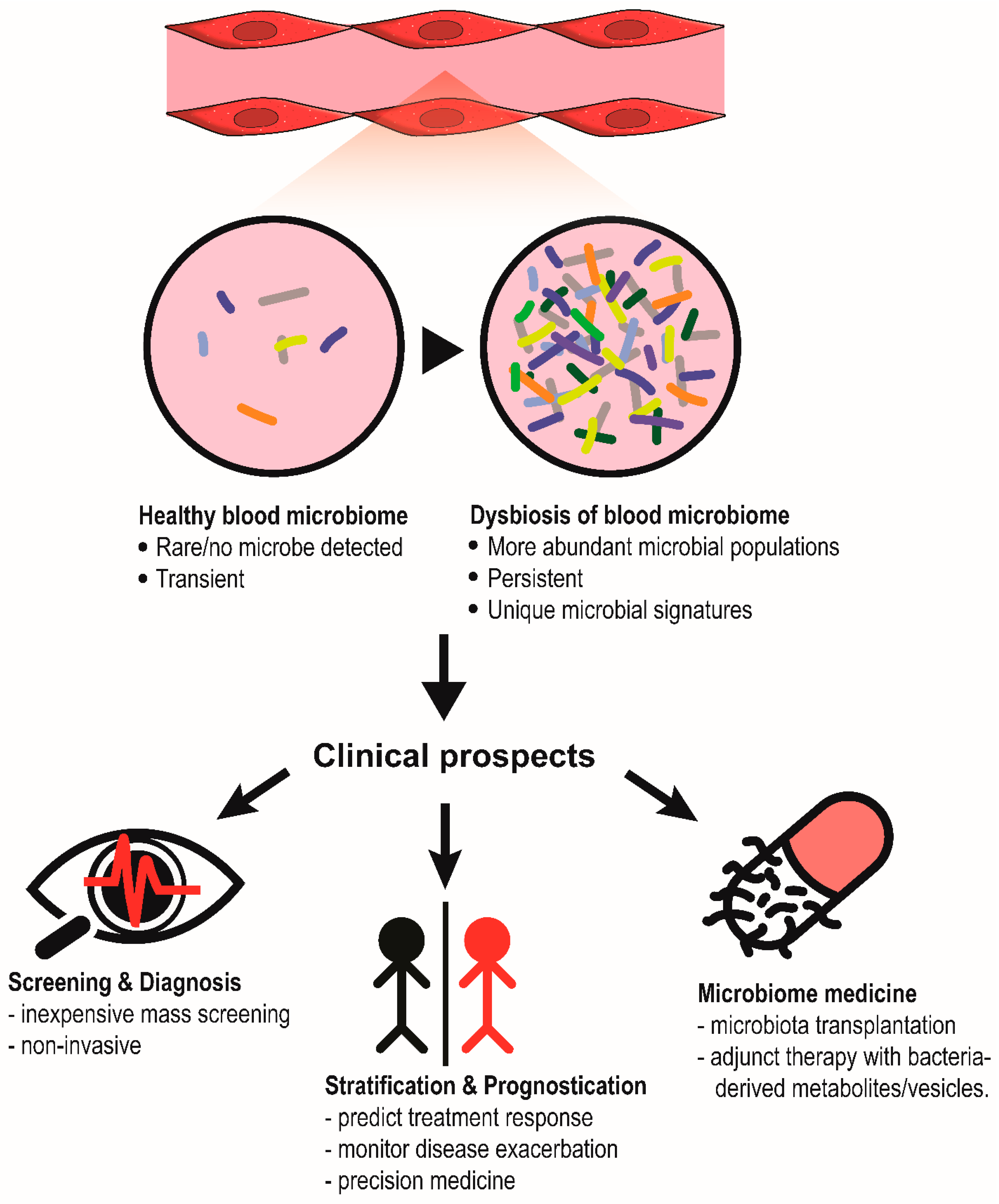{kind=link}
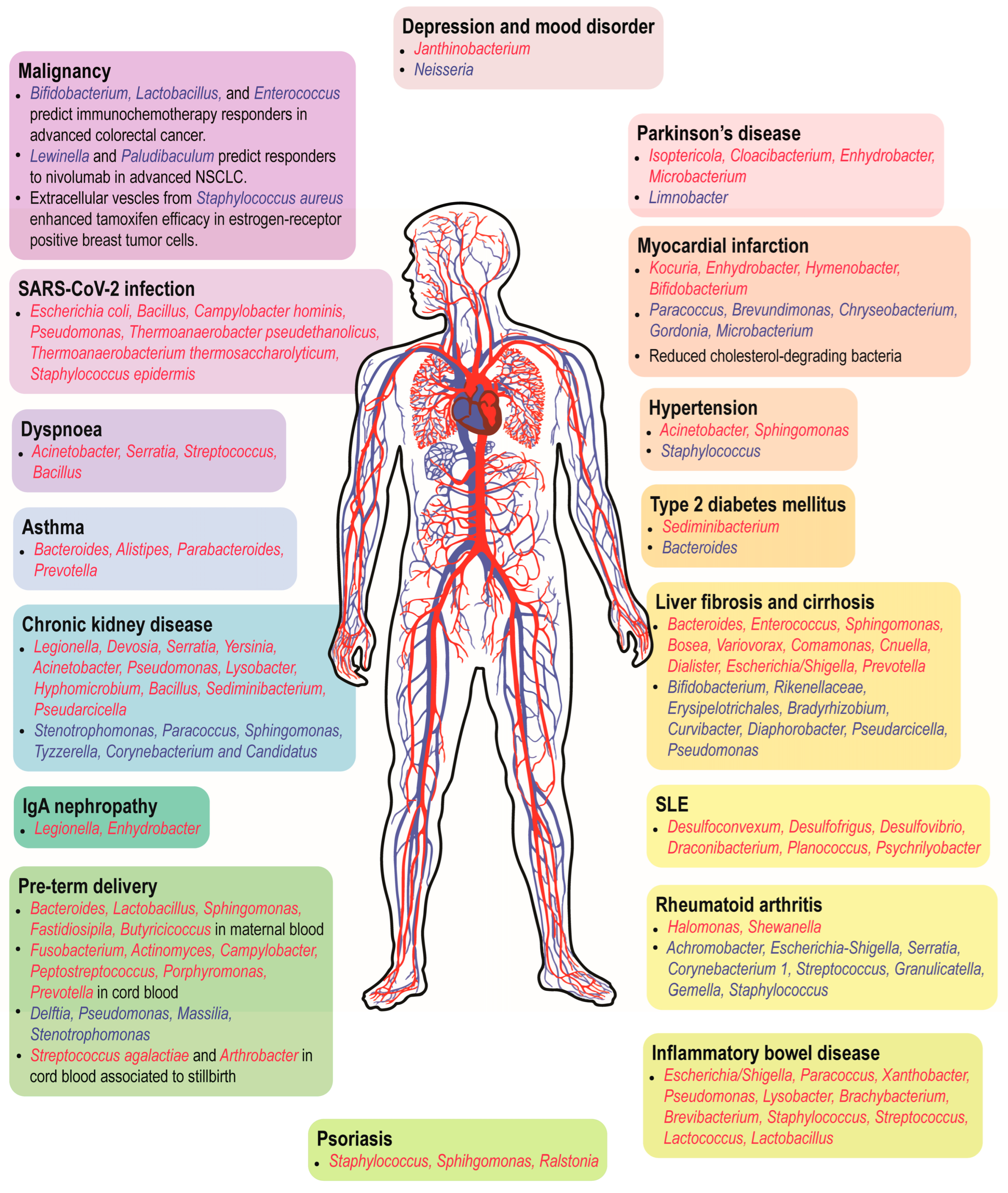{kind=link}
| No. | Disease | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Myocardial infarction | Case-control study | Case = 103; Control = 99 | 16S rDNA (V3-V4 region) sequencing |
| [19] |
| 2. | Myocardial infarction | Case-control study | Case = 29; Control = 29 | 16S rDNA (V3-V4 region) sequencing |
| [20] |
| 3. | Acute (ACS) and chronic coronary syndrome (CCS) | Case-control study | Case (ACS) = 70; Case (CCS) = 70; Control = 70 | 16S rDNA (V3-V4 region) sequencing |
| [21] |
| 4. | Cardiovascular disease (CVD) | Prospective cohort study (D.E.S.I.R.) | CVD = 73; No CVD = 3863 | qPCR of conserved 16S rDNA regions |
| [22] |
| 5. | Cardiovascular mortality | Case-control study (Oslo II) | Case = 227; Control = 178 | 16S rDNA (V3-V5 region) sequencing |
| [23] |
| 6. | T2DM & obesity | Prospective cohort study (D.E.S.I.R.) | T2DM = 131; No T2DM = 3149 | qPCR of conserved 16S rDNA regions |
| [25] |
| 7. | T2DM | Prospective cohort study (135) | T2DM = 50; No T2DM = 100 | 16S rDNA (V5-V6 region) sequencing |
| [26] |
| 8. | Hypertension | Prospective cohort study (135) | Hypertension = 150; No hypertension = 150 | 16S rDNA (V6-V7 region) sequencing |
| [27] |
3.2. Cancers
| No. | Disease | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Pan-cancer | Re-analysis of TCGA whole genome (WGS) and transcriptomic (RNA-seq) sequencing data & cross-sectional patient cohort | TCGA: n = 10,481 (WGS; n = 4831 & RNA-seq; n = 13,285) In-house cohort: HIV- and cancer-free individuals = 69; Prostate cancer = 59; Lung cancer = 25; Melanoma = 16. | WGS, RNAseq and shotgun metagenomic sequencing |
| [28] |
| 2. | Hepatocellular carcinoma (HCC) | Cross-sectional study | HCC = 158; Cirrhosis = 166; Healthy = 402 | 16S rDNA (V3-V4 region) sequencing |
| [29] |
| 3. | Gastric cancer | Case-control study | Gastric cancer = 71; Atypical hyperplasia = 6; Chronic gastritis = 11; Healthy = 13 | 16S rDNA (V1-V2 region) sequencing |
| [30] |
| 4. | Myeloid malignancies | Cross-sectional study | Acute myeloid leukemia (AML) = 612; Myelodysplastic syndromes (MDS) = 640; Myelodysplastic syndromes/myeloproliferative neoplasms (MDS/MPN) = 264; Myeloproliferative neoplasms (MPN) = 354; Healthy = 12. | WGS |
| [31] |
| 5. | Colorectal cancer | Retrospective cohort study | n = 39 | 16S rDNA (V3-V4 region) sequencing |
| [32] |
| 6. | Non-small cell lung cancer (NSCLC) | Single-arm study | n = 72 | 16S rDNA (V3-V4 region) sequencing |
| [33] |
| 7. | Breast cancer | Cross-sectional study | Healthy = 192; Breast cancer = 96 | 16S rDNA (V3-V4 region) sequencing |
| [36] |
3.3. Liver Diseases
| No. | Disease | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Liver fibrosis | Cross-sectional study (FLORINASH) | Spanish cohort: Liver fibrosis = 26; No fibrosis = 11 Italian cohort: Liver fibrosis = 11; No fibrosis = 60 | 16S rDNA (V1-V3 region) sequencing |
| [38] |
| 2. | Cirrhosis | Case-control study | Case = 9; Control = 9 | qPCR of conserved 16S rDNA region |
| [39] |
| 3. | Cirrhosis with or without hepatocellular carcinoma | Case-control study | Case = 66; Control = 14 | 16S rDNA (V3-V4 region) sequencing |
| [40] |
| 4. | Cirrhosis with or without ascites | Case-control study | Case (with ascites) =13; Case (without ascites) =14; Control = 17 | 16S rDNA (V4 region) sequencing |
| [41] |
| 5. | Decompensated liver cirrhosis receiving transjugular intrahepatic portosystemic shunt | Single-arm study | n = 7 | 16S rDNA sequencing |
| [42] |
| 6. | Cirrhosis with portal hypertension | Case-control study | Case = 58; Control = 46 | 16S rDNA (V1-V2 region) sequencing |
| [43] |
| 7. | HCV-induced portal hypertension | Single-arm study | n = 32 | 16S rDNA (V3-V4 region) sequencing |
| [44] |
3.4. Respiratory Diseases
| No. | Disease/Condition | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Asthma | Case-control study | Case = 5; Control = 5 | 16S rDNA (V4 region) sequencing |
| [45] |
| 2. | Asthma | Case-control study | Case = 190; Control = 260 | 16S rDNA (V3-V4 region) sequencing |
| [46] |
| 3. | Smoking | Single-arm from a longitudinal cohort study (COPDGene study) | n = 3655 former and current smokers | RNAseq |
| [47] |
| 4. | Smoking | Case-control study | Case = 20; Control = 21 | 16S rDNA (V4 region) sequencing |
| [49] |
| 5. | COVID-19 | Meta-analysis of public transcriptomic data | 17 PBMC of normal samples; 17 PBMC COVID-19 (GSE152418) | RNAseq |
| [48] |
3.5. Kidney Dysfunction
3.6. Immune and Inflammatory Disorders
| No. | Disease/Condition | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | SLE | Case-control study | Case = 19; control = 30 | 16S rDNA (V4 region) sequencing |
| [49] |
| 2. | SLE | Case-control study | Case = 11; control = 9 | 16S rDNA (V4 region) sequencing |
| [54] |
| 3. | Rheumatoid arthritis | Case-control study | Case = 20; control = 4 | 16S rDNA (V4 region) sequencing |
| [55] |
| 4. | Immunosuppression post liver transplant | Single -arm study | n = 51 | WGS of cell-free DNA |
| [58] |
| 5. | HIV infection | Case-control study | Case = 40; control = 51 | 16S rDNA (V4 region) sequencing |
| [49] |
| 6. | HIV infection | Two cross-sectional studies & one single arm study | 227 HIV-infected patients; 15 healthy individuals | qPCR of conserved 16S rDNA region |
| [59] |
| 7. | Peritonitis | Porcine experiment; pre- and post-fecal induced peritonitis | n = 6 domestic pigs | 16S rDNA (V3-V4 region) sequencing |
| [60] |
| 8. | Inflammatory bowel disease (including Crohn’s disease and ulcerative colitis) | Case-control study | Crohn’s disease = 8; ulcerative colitis = 8; control = 7 | 16S rDNA (V3-V4 region) sequencing |
| [61] |
| 9. | Pancreatitis | Case-control study | Case = 50; control = 12 | 16S rDNA (V3 region) sequencing |
| [62] |
| 10. | Large vessel vasculitis (including giant cell arteritis & Takayasu’s arteritis) | Case-control study | Giant cell arteritis = 11; Takayasu’s arteritis = 20; Healthy = 15. | 16S rDNA (V3-V4 region) sequencing |
| [63] |
3.7. Pregnancy Complications
| No. | Disease/Condition | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Preterm birth | Case control study | Case = 21; Control = 20 | 16S rDNA (V3-V4 region) sequencing |
| [64] |
| 2. | Stillbirth and preterm birth | Case control study | Stillbirth = 60; Preterm birth = 75 Live term (>37 weeks) birth = 101 | 16S rDNA (V4 region) sequencing |
| [65] |
| 3. | Toxoplasma gondii, Others (Hepatitis B virus, Human Papillomavirus [HPV]) Rubella virus, Cytomegalovirus, Herpes simplex virus | Population-based cross-sectional study | n = 107,763 healthy controls | WGS of cell-free DNA |
| [67] |
3.8. Other Health Complications
| No. | Disease | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Rosacea (skin disease) | Case-control study | Case = 10; control = 30 | 16S rDNA (V3-V4 region) sequencing |
| [68] |
| 2. | Psoriasis (skin disease) | Case-control study | Case = 20; control = 8 | 16S rDNA (full length) sequencing |
| [69] |
| 3. | Hidradenitis suppurativa (skin disorder) | Case-control study | Case = 27; control = 26 | 16S rDNA (V3-V4 region) sequencing |
| [70] |
| 4. | Major depression | Case-control study | Case = 56; control = 56 | 16S rDNA (V3-V4 region) sequencing |
| [71] |
| 5. | Parkinson’s disease | Case-control study | Case = 103; Control = 104 | 16S rDNA (V3-V4 region) sequencing |
| [72] |
| 6. | Autism | Case-control study (without statistical analysis) | Case = 15 mother-child pairs; Control = 6 healthy individuals | Culture |
| [73] |
| 7. | Polycystic ovary syndrome | Case-control study | Case = 24; control = 24 | 16S rDNA (V3-V4 region) sequencing |
| [74] |
| 8. | Surgical-induced sepsis | Prospective cohort study | Healthy = 5; Non-infected = 7; Infected = 10; Sepsis = 18; Septic shock = 11 | 16S rDNA (V3 region) sequencing |
| [75] |
| Total parenteral nutrition (TPN) administration | Mouse experiment | Chow-fed: 6; Chow-fed with jugular vein catheter insertion = 6; Intralipid-based TPN = 6; Omegaven-based TPN = 6 | 16S rDNA (V3-V4 region) sequencing |
| [76] |
4. Controversies and Counterclaims
4.1. Susceptibility of Low-Biomass Samples to Exogenous Contamination
4.2. The Blood Microbiome: Viable Colonizers or Cell-Free DNA?
5. Knowledge Gaps and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lian, C.W.K.; Poh, S.E.; Lee, C.K.; Chan, H.M.T.; Yan, G.; Kong, K.W.; Lau, L.; Thomas Lee, W.Y.; Cheng, C.; Hoon, S.; et al. Towards a rapid-turnaround low-depth unbiased metagenomics sequencing workflow on the Illumina platforms. medRxiv 2023. medRxiv:2023.01.02.22283504. [Google Scholar]
- Cheng, W.Y.; Liu, W.X.; Ding, Y.; Wang, G.; Shi, Y.; Chu, E.S.H.; Wong, S.; Sung, J.J.Y.; Yu, J. High Sensitivity of Shotgun Metagenomic Sequencing in Colon Tissue Biopsy by Host DNA Depletion. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Kilian, M.; Chapple, I.L.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.; Tonetti, M.S.; Wade, W.G.; Zaura, E. The oral microbiome—An update for oral healthcare professionals. Br. Dent. J. 2016, 221, 657–666. [Google Scholar] [CrossRef]
- Jones-Freeman, B.; Chonwerawong, M.; Marcelino, V.R.; Deshpande, A.V.; Forster, S.C.; Starkey, M.R. The microbiome and host mucosal interactions in urinary tract diseases. Mucosal Immunol. 2021, 14, 779–792. [Google Scholar] [CrossRef]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Risely, A. Applying the core microbiome to understand host-microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Velmurugan, G.; Dinakaran, V.; Rajendhran, J.; Swaminathan, K. Blood Microbiota and Circulating Microbial Metabolites in Diabetes and Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 835–847. [Google Scholar] [CrossRef]
- Damgaard, C.; Magnussen, K.; Enevold, C.; Nilsson, M.; Tolker-Nielsen, T.; Holmstrup, P.; Nielsen, C.H. Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PLoS ONE 2015, 10, e0120826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aquila, P.; Giacconi, R.; Malavolta, M.; Piacenza, F.; Burkle, A.; Villanueva, M.M.; Dolle, M.E.T.; Jansen, E.; Grune, T.; Gonos, E.S.; et al. Microbiome in Blood Samples From the General Population Recruited in the MARK-AGE Project: A Pilot Study. Front. Microbiol. 2021, 12, 707515. [Google Scholar] [CrossRef] [PubMed]
- Paisse, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Pammi, M.; Thapa, S.; Balderas, M.; Runge, J.K.; Venkatachalam, A.; Luna, R.A. Microbiome signatures in neonatal central line associated bloodstream infections. PLoS ONE 2020, 15, e0227967. [Google Scholar] [CrossRef] [Green Version]
- Scarsella, E.; Sandri, M.; Monego, S.D.; Licastro, D.; Stefanon, B. Blood Microbiome: A New Marker of Gut Microbial Population in Dogs? Vet. Sci. 2020, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Chia, M.; Ko, K.K.; Chen, H.; Liu, J.; Loh, M.; Nagarajan, N. No evidence for a common blood microbiome based on a population study of 9770 healthy humans. bioRxiv 2022. bioRxiv:2022.07.29.502098. [Google Scholar]
- Raeisi, J.; Oloomi, M.; Zolfaghari, M.; Siadat, S.D.; Zargar, M.; Pourramezan, Z. Bacterial DNA Detection in the Blood of Healthy Subjects. Iran. Biomed. J. 2022, 26, 230–239. [Google Scholar] [CrossRef]
- Amar, J.; Lelouvier, B.; Servant, F.; Lluch, J.; Burcelin, R.; Bongard, V.; Elbaz, M. Blood Microbiota Modification After Myocardial Infarction Depends Upon Low-Density Lipoprotein Cholesterol Levels. J. Am. Heart Assoc. 2019, 8, e011797. [Google Scholar] [CrossRef]
- Khan, I.; Khan, I.; Kakakhel, M.A.; Xiaowei, Z.; Ting, M.; Ali, I.; Fei, Y.; Jianye, Z.; Zhiqiang, L.; Lizhe, A. Comparison of Microbial Populations in the Blood of Patients With Myocardial Infarction and Healthy Individuals. Front. Microbiol. 2022, 13, 845038. [Google Scholar] [CrossRef]
- Khan, I.; Khan, I.; Usman, M.; Jianye, Z.; Wei, Z.X.; Ping, X.; Zhiqiang, L.; Lizhe, A. Analysis of the blood bacterial composition of patients with acute coronary syndrome and chronic coronary syndrome. Front. Cell. Infect. Microbiol. 2022, 12, 943808. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.; Midtervoll, I.; Samuelsen, S.O.; Kristoffersen, A.K.; Enersen, M.; Haheim, L.L. The blood microbiome and its association to cardiovascular disease mortality: Case-cohort study. BMC Cardiovasc. Disord. 2022, 22, 344. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Zhou, H.; Jing, Y.; Dong, C. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J. Clin. Lab. Anal. 2019, 33, e22842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Zhou, H.; Lu, H.; Chen, X.; Zhou, L.; Zhang, J.; Wu, J.; Dong, C. Associations Between Peripheral Blood Microbiome and the Risk of Hypertension. Am. J. Hypertens. 2021, 34, 1064–1070. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Cho, E.J.; Leem, S.; Kim, S.A.; Yang, J.; Lee, Y.B.; Kim, S.S.; Cheong, J.Y.; Cho, S.W.; Kim, J.W.; Kim, S.M.; et al. Circulating Microbiota-Based Metagenomic Signature for Detection of Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 7536. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Chen, B.; Pan, H.; Wang, D.; Liu, M.; Yang, Y.; Zou, M.; Yang, J.; Xiao, K.; Zhao, R.; et al. Detection of Microbial 16S rRNA Gene in the Serum of Patients With Gastric Cancer. Front. Oncol. 2019, 9, 608. [Google Scholar] [CrossRef]
- Woerner, J.; Huang, Y.; Hutter, S.; Gurnari, C.; Sanchez, J.M.H.; Wang, J.; Huang, Y.; Schnabel, D.; Aaby, M.; Xu, W.; et al. Circulating microbial content in myeloid malignancy patients is associated with disease subtypes and patient outcomes. Nat. Commun. 2022, 13, 1038. [Google Scholar] [CrossRef]
- Yang, D.; Wang, X.; Zhou, X.; Zhao, J.; Yang, H.; Wang, S.; Morse, M.A.; Wu, J.; Yuan, Y.; Li, S.; et al. Blood microbiota diversity determines response of advanced colorectal cancer to chemotherapy combined with adoptive T cell immunotherapy. Oncoimmunology 2021, 10, 1976953. [Google Scholar] [CrossRef] [PubMed]
- Ouaknine Krief, J.; Helly de Tauriers, P.; Dumenil, C.; Neveux, N.; Dumoulin, J.; Giraud, V.; Labrune, S.; Tisserand, J.; Julie, C.; Emile, J.F.; et al. Role of antibiotic use, plasma citrulline and blood microbiome in advanced non-small cell lung cancer patients treated with nivolumab. J. Immunother. Cancer 2019, 7, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Kwon, H.; Lim, W.; Moon, B.I. Staphylococcus aureus-Derived Extracellular Vesicles Enhance the Efficacy of Endocrine Therapy in Breast Cancer Cells. J. Clin. Med. 2022, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66. [Google Scholar] [CrossRef]
- Lelouvier, B.; Servant, F.; Paisse, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef] [Green Version]
- Traykova, D.; Schneider, B.; Chojkier, M.; Buck, M. Blood Microbiome Quantity and the Hyperdynamic Circulation in Decompensated Cirrhotic Patients. PLoS ONE 2017, 12, e0169310. [Google Scholar] [CrossRef] [Green Version]
- Kajihara, M.; Koido, S.; Kanai, T.; Ito, Z.; Matsumoto, Y.; Takakura, K.; Saruta, M.; Kato, K.; Odamaki, T.; Xiao, J.Z.; et al. Characterisation of blood microbiota in patients with liver cirrhosis. Eur. J. Gastroenterol. Hepatol. 2019, 31, 1577–1583. [Google Scholar] [CrossRef]
- Santiago, A.; Pozuelo, M.; Poca, M.; Gely, C.; Nieto, J.C.; Torras, X.; Roman, E.; Campos, D.; Sarrabayrouse, G.; Vidal, S.; et al. Alteration of the serum microbiome composition in cirrhotic patients with ascites. Sci. Rep. 2016, 6, 25001. [Google Scholar] [CrossRef] [Green Version]
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019, 68, 578–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gedgaudas, R.; Bajaj, J.S.; Skieceviciene, J.; Varkalaite, G.; Jurkeviciute, G.; Gelman, S.; Valantiene, I.; Zykus, R.; Pranculis, A.; Bang, C.; et al. Circulating microbiome in patients with portal hypertension. Gut Microbes 2022, 14, 2029674. [Google Scholar] [CrossRef] [PubMed]
- Virseda-Berdices, A.; Brochado-Kith, O.; Diez, C.; Hontanon, V.; Berenguer, J.; Gonzalez-Garcia, J.; Rojo, D.; Fernandez-Rodriguez, A.; Ibanez-Samaniego, L.; Llop-Herrera, E.; et al. Blood microbiome is associated with changes in portal hypertension after successful direct-acting antiviral therapy in patients with HCV-related cirrhosis. J. Antimicrob. Chemother. 2022, 77, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2018, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Choi, J.P.; Yang, J.; Won, H.K.; Park, C.S.; Song, W.J.; Kwon, H.S.; Kim, T.B.; Kim, Y.K.; Park, H.S.; et al. Metagenome analysis using serum extracellular vesicles identified distinct microbiota in asthmatics. Sci. Rep. 2020, 10, 15125. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Castaldi, P.J.; Chase, R.P.; Yun, J.H.; Lee, S.; Liu, Y.Y.; Hersh, C.P. Peripheral blood microbial signatures in current and former smokers. Sci. Rep. 2021, 11, 19875. [Google Scholar] [CrossRef]
- Dereschuk, K.; Apostol, L.; Ranjan, I.; Chakladar, J.; Li, W.T.; Rajasekaran, M.; Chang, E.Y.; Ongkeko, W.M. Identification of Lung and Blood Microbiota Implicated in COVID-19 Prognosis. Cells 2021, 10, 1452. [Google Scholar] [CrossRef]
- Luo, Z.; Alekseyenko, A.V.; Ogunrinde, E.; Li, M.; Li, Q.Z.; Huang, L.; Tsao, B.P.; Kamen, D.L.; Oates, J.C.; Li, Z.; et al. Rigorous Plasma Microbiome Analysis Method Enables Disease Association Discovery in Clinic. Front. Microbiol. 2020, 11, 613268. [Google Scholar] [CrossRef]
- Shah, N.B.; Allegretti, A.S.; Nigwekar, S.U.; Kalim, S.; Zhao, S.; Lelouvier, B.; Servant, F.; Serena, G.; Thadhani, R.I.; Raj, D.S.; et al. Blood Microbiome Profile in CKD: A Pilot Study. Clin. J. Am. Soc. Nephrol. 2019, 14, 692–701. [Google Scholar] [CrossRef]
- Merino-Ribas, A.; Araujo, R.; Pereira, L.; Campos, J.; Barreiros, L.; Segundo, M.A.; Silva, N.; Costa, C.F.F.A.; Quelhas-Santos, J.; Trindade, F.; et al. Vascular Calcification and the Gut and Blood Microbiome in Chronic Kidney Disease Patients on Peritoneal Dialysis: A Pilot Study. Biomolecules 2022, 12, 867. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.B.; Nigwekar, S.U.; Kalim, S.; Lelouvier, B.; Servant, F.; Dalal, M.; Krinsky, S.; Fasano, A.; Tolkoff-Rubin, N.; Allegretti, A.S. The Gut and Blood Microbiome in IgA Nephropathy and Healthy Controls. Kidney360 2021, 2, 1261. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carrasco, V.; Soriano-Lerma, A.; Soriano, M.; Gutierrez-Fernandez, J.; Garcia-Salcedo, J.A. Urinary Microbiome: Yin and Yang of the Urinary Tract. Front. Cell. Infect. Microbiol. 2021, 11, 617002. [Google Scholar] [CrossRef] [PubMed]
- James, W.A.; Ogunrinde, E.; Wan, Z.; Kamen, D.L.; Oates, J.; Gilkeson, G.S.; Jiang, W. A Distinct Plasma Microbiome but Not Gut Microbiome in Patients with Systemic Lupus Erythematosus Compared to Healthy Individuals. J. Rheumatol. 2022, 49, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Hammad, D.B.M.; Hider, S.L.; Liyanapathirana, V.C.; Tonge, D.P. Molecular Characterization of Circulating Microbiome Signatures in Rheumatoid Arthritis. Front. Cell. Infect. Microbiol. 2019, 9, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audo, R.; Sanchez, P.; Riviere, B.; Mielle, J.; Tan, J.; Lukas, C.; Macia, L.; Morel, J.; Immediato Daien, C. Rheumatoid arthritis is associated with increased gut permeability and bacterial translocation which are reversed by inflammation control. Rheumatology 2022, 62, 1264–1271. [Google Scholar] [CrossRef]
- Bhat, M.; Pasini, E.; Copeland, J.; Angeli, M.; Husain, S.; Kumar, D.; Renner, E.; Teterina, A.; Allard, J.; Guttman, D.S.; et al. Impact of Immunosuppression on the Metagenomic Composition of the Intestinal Microbiome: A Systems Biology Approach to Post-Transplant Diabetes. Sci. Rep. 2017, 7, 10277. [Google Scholar] [CrossRef] [Green Version]
- Okumura, T.; Horiba, K.; Kamei, H.; Takeuchi, S.; Suzuki, T.; Torii, Y.; Kawada, J.I.; Takahashi, Y.; Ogura, Y.; Ogi, T.; et al. Temporal dynamics of the plasma microbiome in recipients at early post-liver transplantation: A retrospective study. BMC Microbiol. 2021, 21, 104. [Google Scholar] [CrossRef]
- Jiang, W.; Lederman, M.M.; Hunt, P.; Sieg, S.F.; Haley, K.; Rodriguez, B.; Landay, A.; Martin, J.; Sinclair, E.; Asher, A.I.; et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J. Infect. Dis. 2009, 199, 1177–1185. [Google Scholar] [CrossRef]
- Hyun, H.; Lee, M.S.; Park, I.; Ko, H.S.; Yun, S.; Jang, D.H.; Kim, S.; Kim, H.; Kang, J.H.; Lee, J.H.; et al. Analysis of Porcine Model of Fecal-Induced Peritonitis Reveals the Tropism of Blood Microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 676650. [Google Scholar] [CrossRef]
- Jones, E.; Stentz, R.; Telatin, A.; Savva, G.M.; Booth, C.; Baker, D.; Rudder, S.; Knight, S.C.; Noble, A.; Carding, S.R. The Origin of Plasma-Derived Bacterial Extracellular Vesicles in Healthy Individuals and Patients with Inflammatory Bowel Disease: A Pilot Study. Genes 2021, 12, 1636. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, C.; Tang, C.; Zhao, X.; He, Q.; Li, J. Identification and Characterization of Blood and Neutrophil-Associated Microbiomes in Patients with Severe Acute Pancreatitis Using Next-Generation Sequencing. Front. Cell. Infect. Microbiol. 2018, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbois, A.C.; Ciocan, D.; Saadoun, D.; Perlemuter, G.; Cacoub, P. Specific microbiome profile in Takayasu’s arteritis and giant cell arteritis. Sci. Rep. 2021, 11, 5926. [Google Scholar] [CrossRef] [PubMed]
- You, Y.A.; Yoo, J.Y.; Kwon, E.J.; Kim, Y.J. Blood microbial communities during pregnancy are associated with preterm birth. Front. Microbiol. 2019, 10, 1122. [Google Scholar] [CrossRef]
- Vander Haar Emilie, L.; Wu, G.; Gyamfi-Bannerman, C.; Thomas, C.; Wapner Ronald, J.; Reddy Uma, M.; Zhao, L.; Silver Robert, M.; Goldenberg Robert, L.; Han Yiping, W. Microbial Analysis of Umbilical Cord Blood Reveals Novel Pathogens Associated with Stillbirth and Early Preterm Birth. mBio 2022, 13, e02036-22. [Google Scholar] [CrossRef]
- Seale, A.C.; Blencowe, H.; Bianchi-Jassir, F.; Embleton, N.; Bassat, Q.; Ordi, J.; Menéndez, C.; Cutland, C.; Briner, C.; Berkley, J.A.; et al. Stillbirth With Group B Streptococcus Disease Worldwide: Systematic Review and Meta-analyses. Clin. Infect. Dis. 2017, 65, S125–S132. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Yu, X.; Du, Y.; Su, F.; Liu, Y.; Li, H.; Liu, Y.; Mu, K.; Liu, Q.; Li, H.; et al. Peripheral Blood Microbiome Analysis via Noninvasive Prenatal Testing Reveals the Complexity of Circulating Microbial Cell-Free DNA. Microbiol. Spectr. 2022, 10, e0041422. [Google Scholar] [CrossRef]
- Yun, Y.; Kim, H.N.; Chang, Y.; Lee, Y.; Ryu, S.; Shin, H.; Kim, W.S.; Kim, H.L.; Nam, J.H. Characterization of the Blood Microbiota in Korean Females with Rosacea. Dermatology 2019, 235, 255–259. [Google Scholar] [CrossRef]
- Chang, C.J.; Zhang, J.; Tsai, Y.L.; Chen, C.B.; Lu, C.W.; Huo, Y.P.; Liou, H.M.; Ji, C.; Chung, W.H. Compositional Features of Distinct Microbiota Base on Serum Extracellular Vesicle Metagenomics Analysis in Moderate to Severe Psoriasis Patients. Cells 2021, 10, 2349. [Google Scholar] [CrossRef]
- Ring, H.C.; Thorsen, J.; Saunte, D.M.; Lilje, B.; Bay, L.; Theut Riis, P.; Larsen, N.; O’Brien Andersen, L.; Vedel Nielsen, H.; Miller, I.M.; et al. Moderate to severe hidradenitis suppurativa patients do not have an altered bacterial composition in peripheral blood compared to healthy controls. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 125–128. [Google Scholar] [CrossRef]
- Ciocan, D.; Cassard, A.M.; Becquemont, L.; Verstuyft, C.; Voican, C.S.; El Asmar, K.; Colle, R.; David, D.; Trabado, S.; Feve, B.; et al. Blood microbiota and metabolomic signature of major depression before and after antidepressant treatment: A prospective case-control study. J. Psychiatry Neurosci. 2021, 46, E358–E368. [Google Scholar] [CrossRef]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Qin, N.; Chen, S.D.; Xiao, Q. Detection of Microbial 16S rRNA Gene in the Blood of Patients With Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Markova, N. Dysbiotic microbiota in autistic children and their mothers: Persistence of fungal and bacterial wall-deficient L-form variants in blood. Sci. Rep. 2019, 9, 13401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, Q.; Zhao, L.; Bin, Y.; Wang, L.; Wang, L.; Zhang, K.; Li, Q. Blood Bacterial 16S rRNA Gene Alterations in Women With Polycystic Ovary Syndrome. Front. Endocrinol. 2022, 13, 814520. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Q.; Tang, C.; Zhao, X.; He, Q.; Tang, X.; Ren, J. Characterization of the blood and neutrophil-specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Immun. Inflamm. Dis. 2021, 9, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, E.; Lou, P.H.; Lemal, P.; Bestmann, L.; Hersberger, M.; Rogler, G.; Kramer, S.D.; Zaugg, M. Gut microbiome and circulating bacterial DNA (“blood microbiome”) in a mouse model of total parenteral nutrition: Evidence of two distinct separate microbiotic compartments. Clin. Nutr. ESPEN 2022, 49, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Chrisman, B.; He, C.; Jung, J.Y.; Stockham, N.; Paskov, K.; Washington, P.; Wall, D.P. The human “contaminome”: Bacterial, viral, and computational contamination in whole genome sequences from 1000 families. Sci. Rep. 2022, 12, 9863. [Google Scholar] [CrossRef]
- Martel, J.; Wu, C.Y.; Huang, P.R.; Cheng, W.Y.; Young, J.D. Pleomorphic bacteria-like structures in human blood represent non-living membrane vesicles and protein particles. Sci. Rep. 2017, 7, 10650. [Google Scholar] [CrossRef] [Green Version]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, K.M.; de Goffau, M.C.; Perez-Munoz, M.E.; Arrieta, M.C.; Backhed, F.; Bork, P.; Braun, T.; Bushman, F.D.; Dore, J.; de Vos, W.M.; et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 2023, 613, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Hornung, B.V.H.; Zwittink, R.D.; Ducarmon, Q.R.; Kuijper, E.J. Response to: ‘Circulating microbiome in blood of different circulatory compartments’ by Schierwagen et al. Gut 2020, 69, 789–790. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jervis-Bardy, J.; Leong, L.E.; Marri, S.; Smith, R.J.; Choo, J.M.; Smith-Vaughan, H.C.; Nosworthy, E.; Morris, P.S.; O’Leary, S.; Rogers, G.B.; et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 2015, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Zrodlowski, T.; Sobonska, J.; Salamon, D.; McFarlane, I.M.; Zietkiewicz, M.; Gosiewski, T. Classical Microbiological Diagnostics of Bacteremia: Are the Negative Results Really Negative? What is the Laboratory Result Telling Us About the “Gold Standard”? Microorganisms 2020, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Panaiotov, S.; Hodzhev, Y.; Tsafarova, B.; Tolchkov, V.; Kalfin, R. Culturable and Non-Culturable Blood Microbiota of Healthy Individuals. Microorganisms 2021, 9, 1464. [Google Scholar] [CrossRef]
- Batani, G.; Bayer, K.; Boge, J.; Hentschel, U.; Thomas, T. Fluorescence in situ hybridization (FISH) and cell sorting of living bacteria. Sci. Rep. 2019, 9, 18618. [Google Scholar] [CrossRef] [Green Version]
- Blattman, S.B.; Jiang, W.; Oikonomou, P.; Tavazoie, S. Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat. Microbiol. 2020, 5, 1192–1201. [Google Scholar] [CrossRef]
- Kuchina, A.; Brettner, L.M.; Paleologu, L.; Roco, C.M.; Rosenberg, A.B.; Carignano, A.; Kibler, R.; Hirano, M.; DePaolo, R.W.; Seelig, G. Microbial single-cell RNA sequencing by split-pool barcoding. Science 2021, 371, eaba5257. [Google Scholar] [CrossRef]
- Homberger, C.; Hayward, R.J.; Barquist, L.; Vogel, J. Improved bacterial single-cell RNA-seq through automated MATQ-seq and Cas9-based removal of rRNA reads. bioRxiv 2022. bioRxiv:2022.11.28.518171. [Google Scholar] [CrossRef]
- Ma, P.; Amemiya, H.M.; He, L.L.; Gandhi, S.J.; Nicol, R.; Bhattacharyya, R.P.; Smillie, C.S.; Hung, D.T. Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell 2023, 186, 877–891. [Google Scholar] [CrossRef] [PubMed]
- Filyk, H.A.; Osborne, L.C. The Multibiome: The Intestinal Ecosystem’s Influence on Immune Homeostasis, Health, and Disease. EBioMedicine 2016, 13, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac Aogain, M.; Narayana, J.K.; Tiew, P.Y.; Ali, N.; Yong, V.F.L.; Jaggi, T.K.; Lim, A.Y.H.; Keir, H.R.; Dicker, A.J.; Thng, K.X.; et al. Integrative microbiomics in bronchiectasis exacerbations. Nat. Med. 2021, 27, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Barrila, J.; Crabbe, A.; Yang, J.; Franco, K.; Nydam, S.D.; Forsyth, R.J.; Davis, R.R.; Gangaraju, S.; Ott, C.M.; Coyne, C.B.; et al. Modeling Host-Pathogen Interactions in the Context of the Microenvironment: Three-Dimensional Cell Culture Comes of Age. Infect. Immun. 2018, 86, e00282-18. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 2018, 9, 1534. [Google Scholar] [CrossRef] [Green Version]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef] [Green Version]


| No. | Disease | Study Design | Sample Size | Detection Method | Findings | Reference |
|---|---|---|---|---|---|---|
| 1. | Chronic kidney disease (CKD) | Cross-sectional study | CKD = 20; Healthy = 20 | 16S rDNA (V3-V4 region) sequencing |
| [50] |
| 2. | CKD patients on peritoneal dialysis (PD) with vascular calcification (VC) | Cross-sectional study | CKD-PD without VC = 12; CKD-PD with VC = 32 | 16S rDNA (V3-V4 region) sequencing |
| [51] |
| 3. | IgA nephropathy (Berger’s disease) | Case-control study | Case = 20; Controls = 20 | 16S rDNA (V3-V4 region) sequencing |
| [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.S.; Tan, S.P.; Wong, D.M.K.; Koo, W.L.Y.; Wong, S.H.; Tan, N.S. The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges. Int. J. Mol. Sci. 2023, 24, 5633. https://doi.org/10.3390/ijms24065633
Cheng HS, Tan SP, Wong DMK, Koo WLY, Wong SH, Tan NS. The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges. International Journal of Molecular Sciences. 2023; 24(6):5633. https://doi.org/10.3390/ijms24065633
Chicago/Turabian StyleCheng, Hong Sheng, Sin Pei Tan, David Meng Kit Wong, Wei Ling Yolanda Koo, Sunny Hei Wong, and Nguan Soon Tan. 2023. "The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges" International Journal of Molecular Sciences 24, no. 6: 5633. https://doi.org/10.3390/ijms24065633