
Synergistic Interaction of the Class IIa HDAC Inhibitor CHDI0039 with Bortezomib in Head and Neck Cancer Cells
 , , and
, , and
Abstract
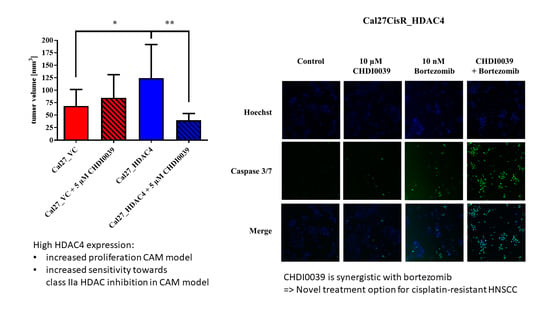
:
1. Introduction
2. Results
2.1. Establishment of a Cellular Test System to Study the Role of Class IIa HDACs in Head and Neck Cancer

2.2. Effect of HDAC4 Overexpression on Cell Proliferation and Tumor Growth
2.3. Transcriptome Changes Induced by HDAC4 Overexpression and Chemical Inhibition of Class IIa HDACs

2.4. Effects of HDAC4 and HDAC5 on Cytotoxicity toward HDACi, Cisplatin and Bortezomib
2.5. Effects of Class IIa HDACi on Cisplatin Induced Cytotoxicity
2.6. Combining Class IIa HDACi with Bortezomib Provides Synergistic Cytotoxicity in Head and Neck Cancer
2.7. CHDI0039 and Bortezomib Synergistically Increase Caspase 3/7 Activation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. MTT Cell Viability Assay
4.4. Synergy Studies According to Chou Talalay
4.5. Caspase 3/7 Activation Assay
4.6. Generation of Cell Lines Stably Expressing HDAC4, HDAC5 or Vector Control
4.7. Chicken Chorioallantoic Membrane (CAM) Model
4.8. RNA Extraction and RNA Sequencing Analysis
4.9. Analysis of RNA Sequencing Data
4.10. Immunoblotting
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorg. Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Hideshima, T.; Anderson, K.C. Histone deacetylase inhibitors in multiple myeloma: From bench to bedside. Int. J. Hematol. 2016, 104, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenet. 2018, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, A.A.J.; Hoffmann, M.J.; Beck, M.L.C.; Poschmann, G.; Petzsch, P.; Wiek, C.; Stühler, K.; Köhrer, K.; Schulz, W.A.; Niegisch, G. HDAC5 expression in urothelial carcinoma cell lines inhibits long-term proliferation but can promote epitheli-al-to-mesenchymal transition. Int. J. Mol. Sci. 2019, 20, 2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaety, M.E.; Gries, A.; Badie, A.; Venkatasamy, A.; Romain, B.; Orvain, C.; Yanagihara, K.; Okamoto, K.; Jung, A.C.; Mellitzer, G.; et al. HDAC4 levels control sensibility toward cisplatin in gastric cancer via the p53-p73/BIK pathway. Cancers 2019, 11, 1747. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.S.; Yang, X.Z.; Wen, Y.F.; Mai, S.J.; Wang, M.H.; Zhang, M.Y.; Steven Zheng, X.F.; Wang, H.Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging 2016, 8, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Suzuki, R.; Ohguchi, H.; Yoshida, Y.; Lu, D.; Cottini, F.; Jakubikova, J.; Bianchi, G.; Harada, T.; Gorgun, G.; et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia 2015, 29, 1918–1927. [Google Scholar] [CrossRef]
- Usami, M.; Kikuchi, S.; Takada, K.; Ono, M.; Sugama, Y.; Arihara, Y.; Hayasaka, N.; Nakamura, H.; Ikeda, Y.; Hirakawa, M.; et al. FOXO3a Activation by HDAC Class IIa Inhibition Induces Cell Cycle Arrest in Pancreatic Cancer Cells. Pancreas 2020, 49, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zheng, J.M.; Chen, J.K.; Yan, X.L.; Chen, H.M.; Nong, W.X.; Huang, H.Q. Histone deacetylase 5 promotes the proliferation of glioma cells by upregulation of Notch 1. Mol. Med. Rep. 2014, 10, 2045–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [Green Version]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: From important roles in differentiation to possible implications in tumourigenesis. J. Cell. Mol. Med. 2011, 15, 1833–1846. [Google Scholar] [CrossRef] [Green Version]
- Autin, P.; Blanquart, C.; Fradin, D. Epigenetic Drugs for Cancer and microRNAs: A Focus on Histone Deacetylase Inhibitors. Cancers 2019, 11, 1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052. [Google Scholar] [CrossRef] [Green Version]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 1113. [Google Scholar] [CrossRef]
- Krieger, V.; Hamacher, A.; Cao, F.; Stenzel, K.; Gertzen, C.G.W.; Schäker-Hübner, L.; Kurz, T.; Gohlke, H.; Dekker, F.J.; Kassack, M.U.; et al. Synthesis of Peptoid-Based Class I-Selective Histone Deacetylase Inhibitors with Chemosensitizing Properties. J. Med. Chem. 2019, 62, 11260–11279. [Google Scholar] [CrossRef]
- Liu, L.; Dong, L.; Bourguet, E.; Fairlie, D.P. Targeting Class IIa HDACs: Insights from Phenotypes and Inhibitors. Curr. Med. Chem. 2021, 28, 8628–8672. [Google Scholar] [CrossRef]
- Morel, D.; Jeffrey, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Bollmann, L.M.; Skerhut, A.J.; Asfaha, Y.; Horstick, N.; Hanenberg, H.; Hamacher, A.; Kurz, T.; Kassack, M.U. The Novel Class IIa Selective Histone Deacetylase Inhibitor YAK540 Is Synergistic with Bortezomib in Leukemia Cell Lines. Int. J. Mol. Sci. 2022, 23, 13398. [Google Scholar] [CrossRef]
- Gosepath, E.M.; Eckstein, N.; Hamacher, A.; Servan, K.; von Jonquieres, G.; Lage, H.; Györffy, B.; Royer, H.D.; Kassack, M.U. Acquired cisplatin resistance in the head–neck cancer cell line Cal27 is associated with decreased DKK1 expression and can partially be reversed by overexpression of DKK1. Int. J. Cancer 2008, 123, 2013–2019. [Google Scholar] [CrossRef]
- Sinatra, L.; Bandolik, J.J.; Roatsch, M.; Sönnichsen, M.; Schoeder, C.T.; Hamacher, A.; Schöler, A.; Borkhardt, A.; Meiler, J.; Bhatia, S.; et al. Hydroxamic Acids Immobilized on Resins (HAIRs): Synthesis of Dual-Targeting HDAC Inhibitors and HDAC Degraders (PROTACs). Angew. Chem. Int. Ed. 2020, 59, 22494–22499. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Lange, F.; Wang, C.; Bandolik, J.J.; Hamacher, A.; Kassack, M.U.; Kurz, T. Novel alkoxyamide-based histone deacetylase inhibitors reverse cisplatin resistance in chemoresistant cancer cells. Bioorg. Med. Chem. 2020, 28, 115108. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Kim, Y.S.; Kim, H.J.; Kim, D.H.; Won, H.R.; Kim, Y.S.; Kim, C.H. HDAC4 degradation by combined TRAIL and valproic acid treatment induces apoptotic cell death of TRAIL-resistant head and neck cancer cells. Sci. Rep. 2018, 8, 12520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Yang, J.; Li, S.W.; Huang, G.; Li, C.; Min, W.P.; Sang, Y. HDAC4 promotes nasopharyngeal carcinoma progression and serves as a therapeutic target. Cell Death Dis. 2021, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Luckhurst, C.A.; Aziz, O.; Beaumont, V.; Bürli, R.W.; Breccia, P.; Maillard, M.C.; Haughan, A.F.; Lamers, M.; Leonard, P.; Matthews, K.L.; et al. Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2019, 29, 83–88. [Google Scholar] [CrossRef]
- Cao, K.; Wang, H.; Fang, Y.; Wang, Y.; Wei, L.; Chen, X.; Jiang, Z.; Wei, X.; Hu, Y. Histone Deacetylase 4 Promotes Osteosarcoma Cell Proliferation and Invasion by Regulating Expression of Proliferating Cell Nuclear Antigen. Front. Oncol. 2019, 9, 870. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Tian, Y.; Jiang, D.; Song, Y. Long noncoding RNA RGMB-AS1 acts as a microRNA-574 sponge thereby enhancing the aggressiveness of gastric cancer via HDAC4 upregulation. Onco. Targets. Ther. 2020, 13, 1691–1704. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.Y.; Xu, T.T.; Wang, Y.; Chang, J.J.; Li, J.; Chen, X.Y.; Chan, X.; Yin, Y.F.; Ni, X.J. Histone deacetylase HDAC4 promotes the proliferation and invasion of glioma cells. Int. J. Oncol. 2018, 53, 2758–2768. [Google Scholar] [CrossRef] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Dong, L.; Zhang, R.; Ying, K.; Shen, H. Transgelin overexpression in lung adenocarcinoma is associated with tumor progression. Int. J. Mol. Med. 2014, 34, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Han, Z.; Wang, X.; Zheng, X.; Wang, S.; Cui, Y. H2B gene family: A prognostic biomarker and correlates with immune infiltration in glioma. Front. Oncol. 2022, 12, 966817. [Google Scholar] [CrossRef]
- Harris, N.L.E.; Vennin, C.; Conway, J.R.W.; Vine, K.L.; Pinese, M.; Cowley, M.J.; Shearer, R.F.; Lucas, M.C.; Herrmann, D.; Allam, A.H.; et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 2017, 36, 4288–4298. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Li, H.; Huang, Q.; Chen, D.; Han, J.; Wang, L.; Pan, C.; Chen, W.; House, M.G.; Nephew, K.P.; et al. SERPINB2 down-regulation contributes to chemoresistance in head and neck cancer. Mol. Carcinog. 2014, 53, 777–786. [Google Scholar] [CrossRef]
- Huang, X.-P.; Li, X.; Situ, M.-Y.; Huang, L.-Y.; Wang, J.-Y.; He, T.-C.; Yan, Q.-H.; Xie, X.-Y.; Zhang, Y.-J.; Gao, Y.-H.; et al. Entinostat reverses cisplatin resistance in esophageal squamous cell carcinoma via down-regulation of multidrug resistance gene 1. Cancer Lett. 2018, 414, 294–300. [Google Scholar] [CrossRef]
- Alves Avelar, L.A.; Schrenk, C.; Sönnichsen, M.; Hamacher, A.; Hansen, F.K.; Schliehe-Diecks, J.; Borkhardt, A.; Bhatia, S.; Kassack, M.U.; Kurz, T. Synergistic induction of apoptosis in resistant head and neck carcinoma and leukemia by alkoxyamide-based histone deacetylase inhibitors. Eur. J. Med. Chem. 2021, 211, 113095. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Kang, D.O.; Na, Y.J.; Yoon, S.; Choi, W.S.; Kang, K.W.; Chung, H.Y.; Jung, J.H.; Min, D.S.; Kim, H.S. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett. 2012, 325, 189–199. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.H.; Chang, Y.F.; Lee, M.S.; Wen, B.C.; Ko, J.C.; Liang, S.K.; Liang, M.C. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer 2016, 16, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelke, L.H.; Hamacher, A.; Proksch, P.; Kassack, M.U. Ellagic acid and resveratrol prevent the development of cisplatin resistance in the epithelial ovarian cancer cell line A2780. J. Cancer 2016, 7, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Prieto, C.; Barrios, D. RaNA-Seq: Interactive RNA-Seq analysis from FASTQ files to functional analysis. Bioinformatics 2019, 36, 1955–1956. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line Cal27 | No. Genes FDR 1: p ≤ 0.05 Fold Change ≥ 2 | No. Genes Bonferroni: p ≤ 0.05 Fold Change ≥ 2 |
|---|---|---|
| Cal27_HDAC4 compared to Cal27_VC | Total: 588 167 up in HDAC4 421 down in HDAC4 | Total: 363 85 up in HDAC4 278 down in HDAC4 |
| Cal27_HDAC4 + 5 µM CHDI0039 compared to Cal27_HDAC4 | Total: 319 133 up in HDAC4 + CHDI0039 186 down in HDAC4 + CHDI0039 | Total: 141 61 up in HDAC4 + CHDI0039 80 down in HDAC4 + CHDI0039 |
| (a) | ||||
|---|---|---|---|---|
| No. | Gene ID | Bonferroni from RNAseq | Fold Difference | Log Rank p-Value from KM Plotter |
| 1 | SERPINB2 | 1.16 × 10−65 | 3.47 | 0.035 |
| 2 | NFATC2 | 1.15 × 10−23 | 3.38 | 0.013 |
| 3 | PLA2G4D | 9.68 × 10−7 | 2.66 | 0.037 |
| 4 | CYTH4 | 1.87 × 10−3 | 2.56 | 0.027 |
| 5 | L3MBTL1 | 1.31 × 10−2 | 2.11 | 0.016 |
| (b) | ||||
| No. | Gene ID | Bonferroni from RNAseq | Fold Difference | Log Rank p-Value from KM Plotter |
| 1 | IGSF1 | 3.02 × 10−2 | −2.02 | 0.021 |
| 2 | HIST1H2BD | 2.07 × 10−7 | −2.10 | 0.028 |
| 3 | ACTBL2 | 3.31 × 10−4 | −2.13 | 0.027 |
| 4 | MSRB3 | 3.43 × 10−14 | −2.20 | 0.020 |
| 5 | PIGR | 7.53 × 10−27 | −3.10 | 0.045 |
| 6 | TAGLN | 7.59 × 10−10 | −3.93 | 0.010 |
| 7 | LRRC4 | 3.39 × 10−7 | −4.03 | 0.001 |
| 8 | CAMK2A | 6.92 × 10−7 | −4.24 | 0.001 |
| Compound | Cal27 Clones | Cal27CisR Clones | ||||
|---|---|---|---|---|---|---|
| VC | HDAC4 | HDAC5 | VC | HDAC4 | HDAC5 | |
| Cisplatin [µM] | 2.77 ± 0.20 | 2.47 ± 0.20 | 3.28 ± 0.21 | 11.0 ± 1.85 | 13.7 ± 1.77 | 11.3 ± 1.41 |
| Vorinostat [µM] | 1.33 ± 0.08 | 1.49 ± 0.10 | 0.92 ± 0.06 | 2.12 ± 0.13 | 2.04 ± 0.09 | 2.58 ± 0.20 |
| CHDI0039 [µM] | 9.69 ± 0.62 | 12.8 ± 0.53 | 14.1 ± 0.61 | 22.6 ± 1.11 | 24.7 ± 1.39 | 24.5 ± 2.19 |
| Bortezomib [µM] | 0.009 ± 0.0002 | 0.005 ± 0.0002 | 0.005 ± 0.0004 | 0.025 ± 0.003 | 0.020 ± 0.003 | 0.022 ± 0.003 |
| Cal27CisR | ||||||
|---|---|---|---|---|---|---|
| VC | HDAC4 | HDAC5 | ||||
| Cisplatin [µM] | ||||||
| IC50 | SF | IC50 | SF | IC50 | SF | |
| Control | 51.3 | - | 49.9 | - | 46.1 | - |
| +2.5 µM CHDI0039 | 48.9 | 1.05 (ns) | 45.8 | 1.09 (ns) | 54.6 | 0.84 (ns) |
| +5.0 µM CHDI0039 | 32.5 | 1.58 (***) | 88.7 | 0.56 (***) | 39.5 | 1.17 (ns) |
| +0.75 µM Vorinostat | 3.18 | 16.1 (***) | 3.01 | 16.6 (***) | 2.60 | 17.7 (***) |
| Cal27CisR | |||||||
|---|---|---|---|---|---|---|---|
| VC | HDAC4 | HDAC5 | |||||
| Bortezomib [nM] | |||||||
| IC50 | SF | IC50 | SF | IC50 | SF | ||
| 25.3 | - | 20.4 | - | 21.6 | - | ||
| CHDI0039 [µM] | +2.5 | 22.1 | 1.14 | 9.62 | 2.12 † | 11.0 | 1.96 † |
| +5.0 | 9.62 | 2.63 † | 7.91 | 2.58 † | 8.74 | 2.47 † | |
| +7.5 | 8.48 | 2.98 † | 6.99 | 2.92 † | 8.03 | 2.69 † | |
| +10 | 8.88 | 2.85 † | 7.43 | 2.75 † | 7.74 | 2.79 † | |
| (a) | ||||
| Cal27CisR_VC | ||||
| Bortezomib [nM] | ||||
| 10 | 15 | 20 | ||
| CHDI0039 [µM] | +2.5 | * | 0.51 | 0.61 |
| +5.0 | 0.69 | 0.54 | 0.68 | |
| +7.5 | 0.58 | 0.60 | 0.73 | |
| +10 | 0.63 | 0.66 | 0.78 | |
| (b) | ||||
| Cal27CisR_HDAC4 | ||||
| Bortezomib [nM] | ||||
| 10 | 15 | 20 | ||
| CHDI0039 [µM] | +2.5 | 0.76 | 0.53 | 0.67 |
| +5.0 | 0.55 | 0.59 | 0.74 | |
| +7.5 | 0.52 | 0.64 | 0.79 | |
| +10 | 0.57 | 0.70 | 0.85 | |
| (c) | ||||
| Cal27CisR_HDAC5 | ||||
| Bortezomib [nM] | ||||
| 10 | 15 | 20 | ||
| CHDI0039 [µM] | +2.5 | 0.59 | 0.50 | 0.62 |
| +5.0 | 0.47 | 0.56 | 0.68 | |
| +7.5 | 0.48 | 0.62 | 0.75 | |
| +10 | 0.55 | 0.67 | 0.81 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schrenk, C.; Bollmann, L.M.; Haist, C.; Bister, A.; Wiek, C.; Wecker, M.; Roth, D.; Petzsch, P.; Köhrer, K.; Hamacher, A.; et al. Synergistic Interaction of the Class IIa HDAC Inhibitor CHDI0039 with Bortezomib in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2023, 24, 5553. https://doi.org/10.3390/ijms24065553
Schrenk C, Bollmann LM, Haist C, Bister A, Wiek C, Wecker M, Roth D, Petzsch P, Köhrer K, Hamacher A, et al. Synergistic Interaction of the Class IIa HDAC Inhibitor CHDI0039 with Bortezomib in Head and Neck Cancer Cells. International Journal of Molecular Sciences. 2023; 24(6):5553. https://doi.org/10.3390/ijms24065553
Chicago/Turabian StyleSchrenk, Christian, Lukas M. Bollmann, Corinna Haist, Arthur Bister, Constanze Wiek, Maria Wecker, Dennis Roth, Patrick Petzsch, Karl Köhrer, Alexandra Hamacher, and et al. 2023. "Synergistic Interaction of the Class IIa HDAC Inhibitor CHDI0039 with Bortezomib in Head and Neck Cancer Cells" International Journal of Molecular Sciences 24, no. 6: 5553. https://doi.org/10.3390/ijms24065553