
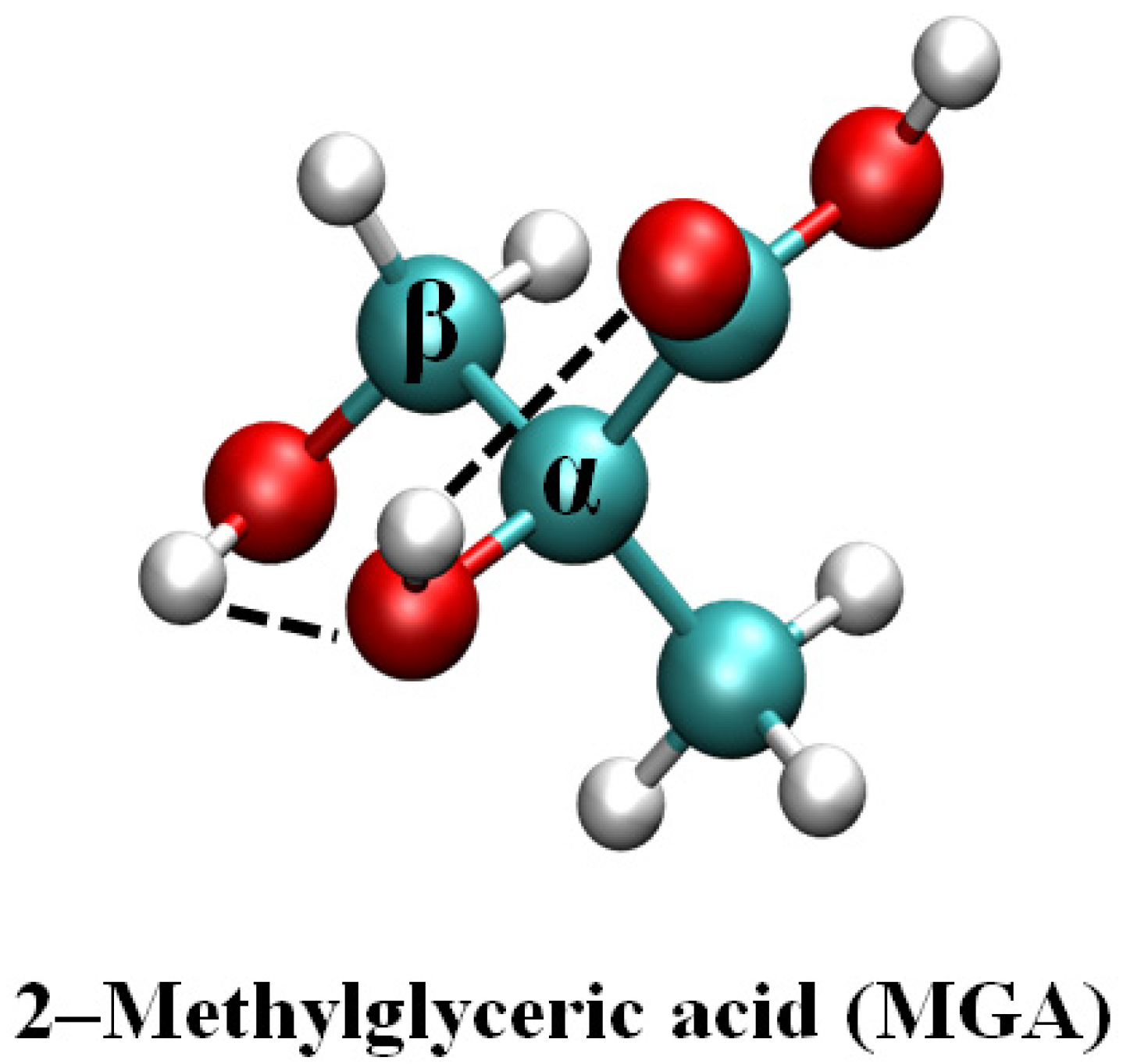
Theoretical Study on the Gas-Phase and Aqueous Interface Reaction Mechanism of Criegee Intermediates with 2-Methylglyceric Acid and the Nucleation of Products
Abstract
:1. Introduction
2. Results and Discussion
2.1. Gas-Phase Reactions
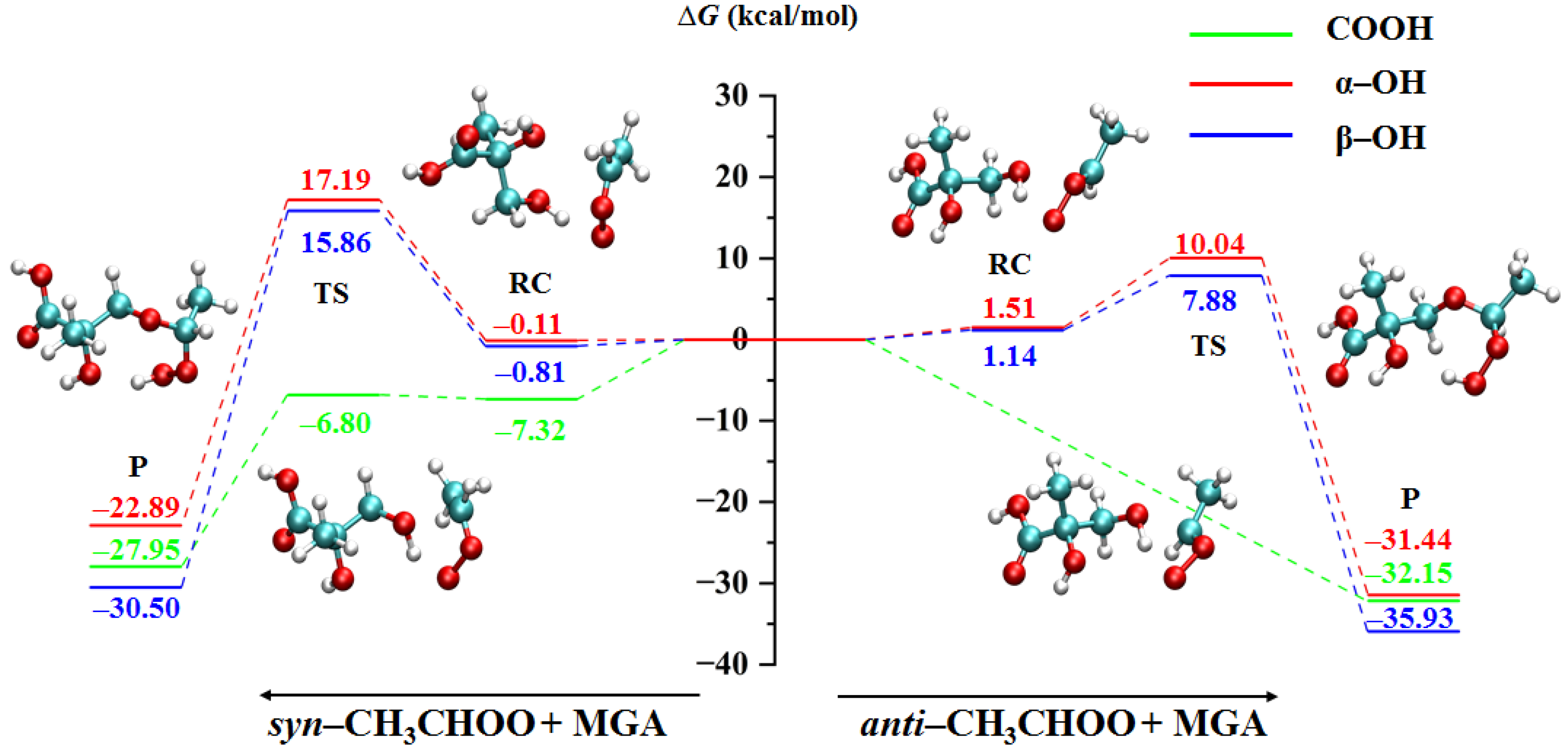
2.1.1. Bimolecular Reactions
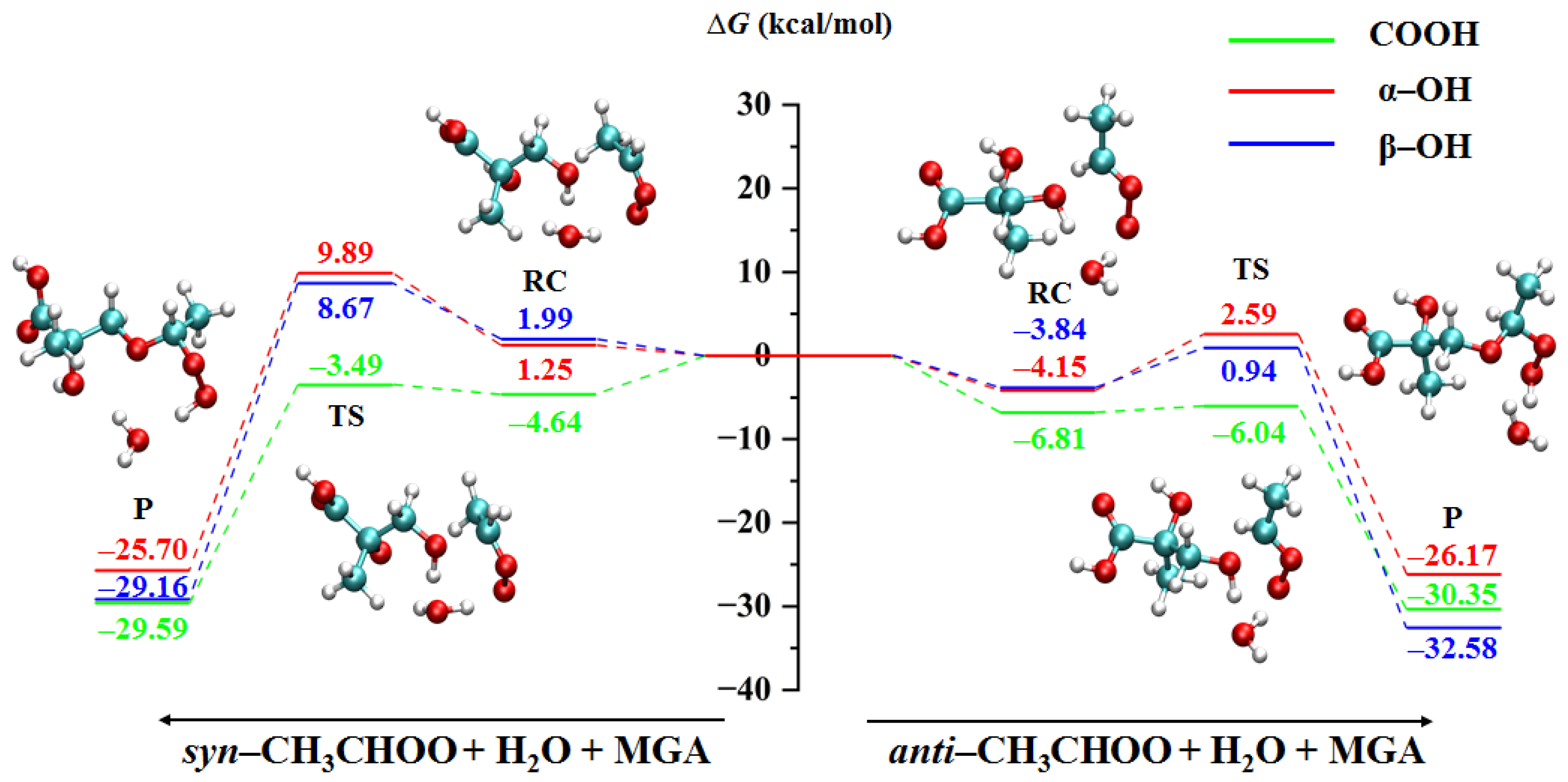
2.1.2. Water-Mediated Reactions
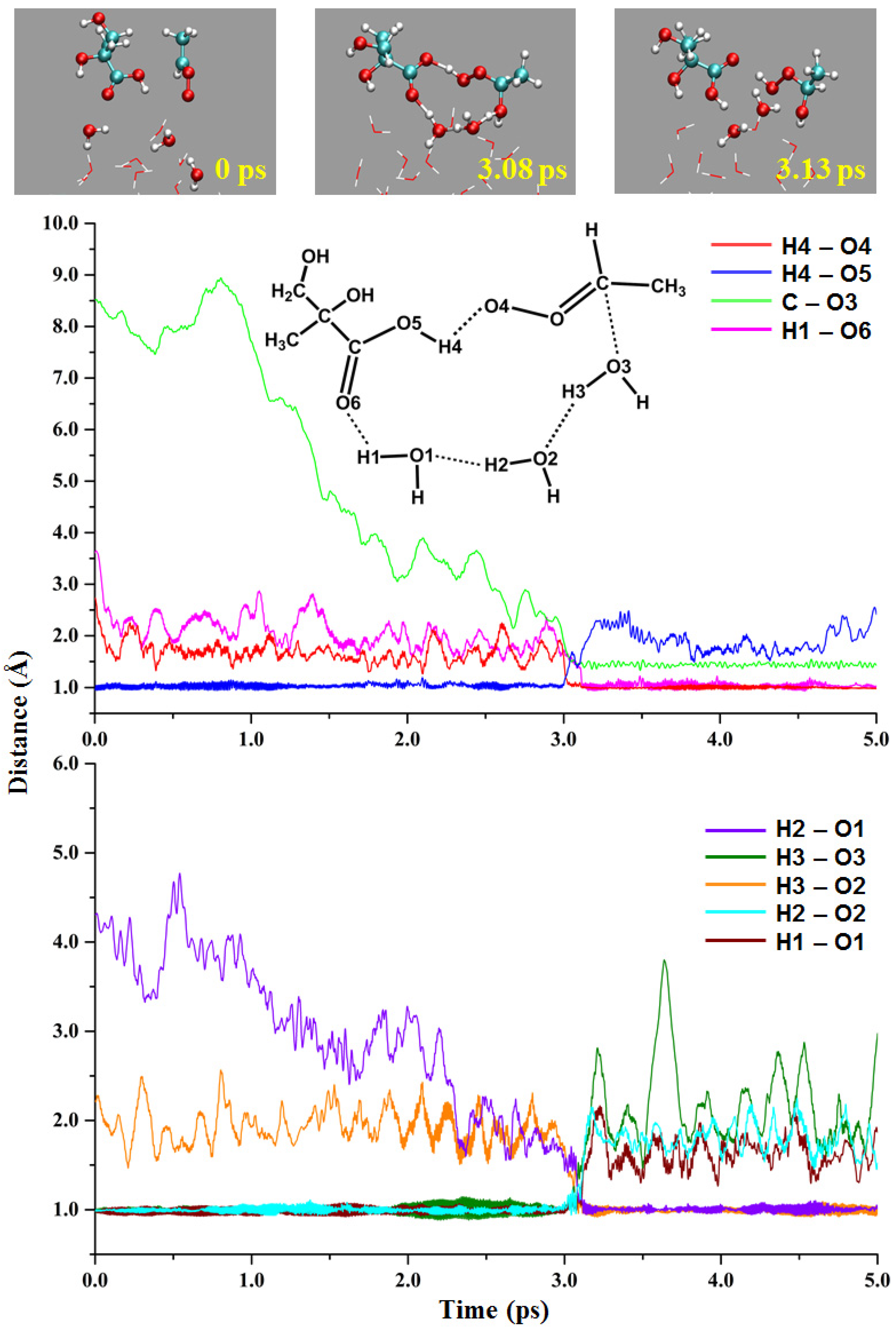
2.2. Gas-Liquid Interface Reactions
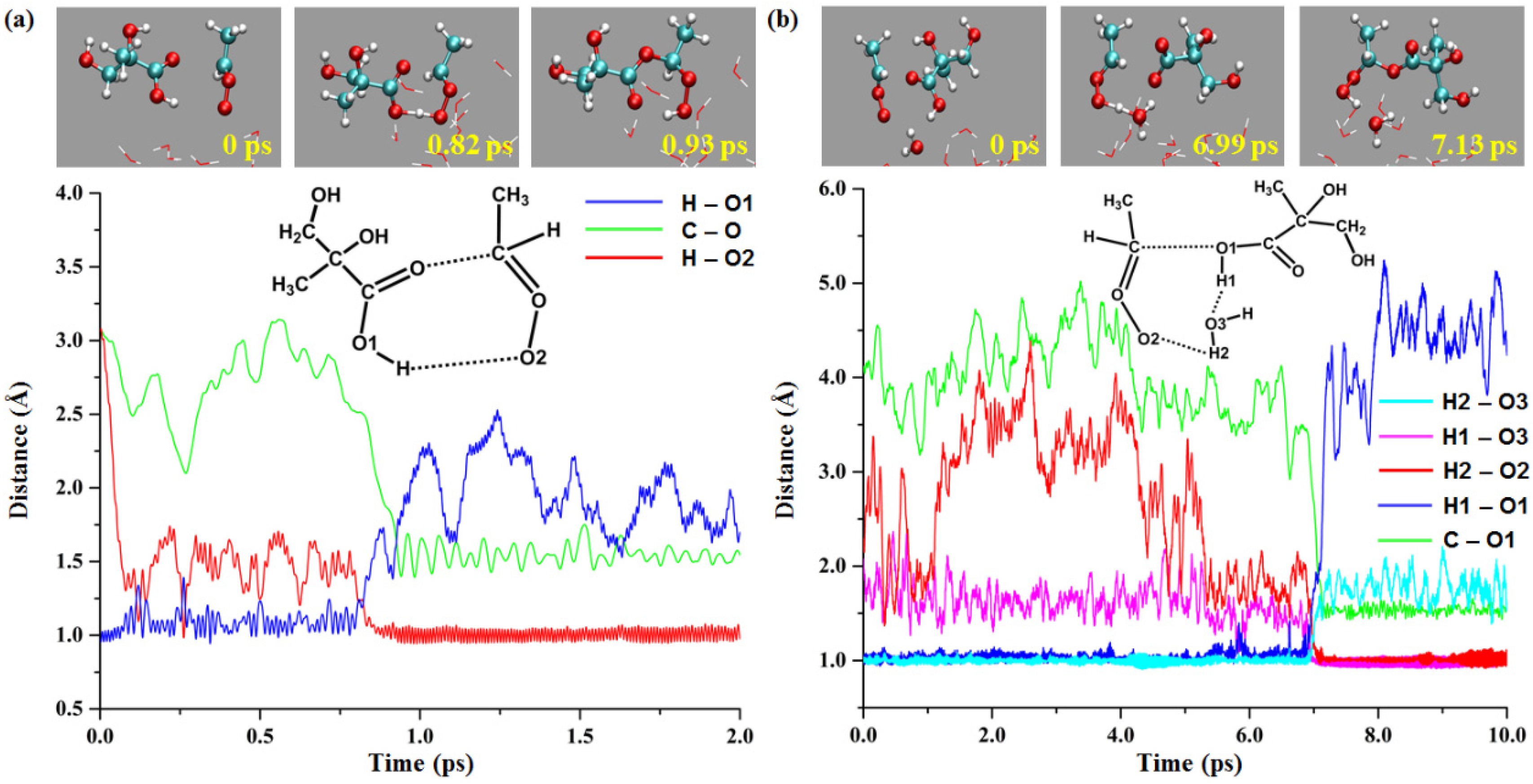
2.2.1. Reaction of Anti-CH3CHOO with the MGA COOH Group
2.2.2. MGA-Mediated Hydration of Anti-CH3CHOO
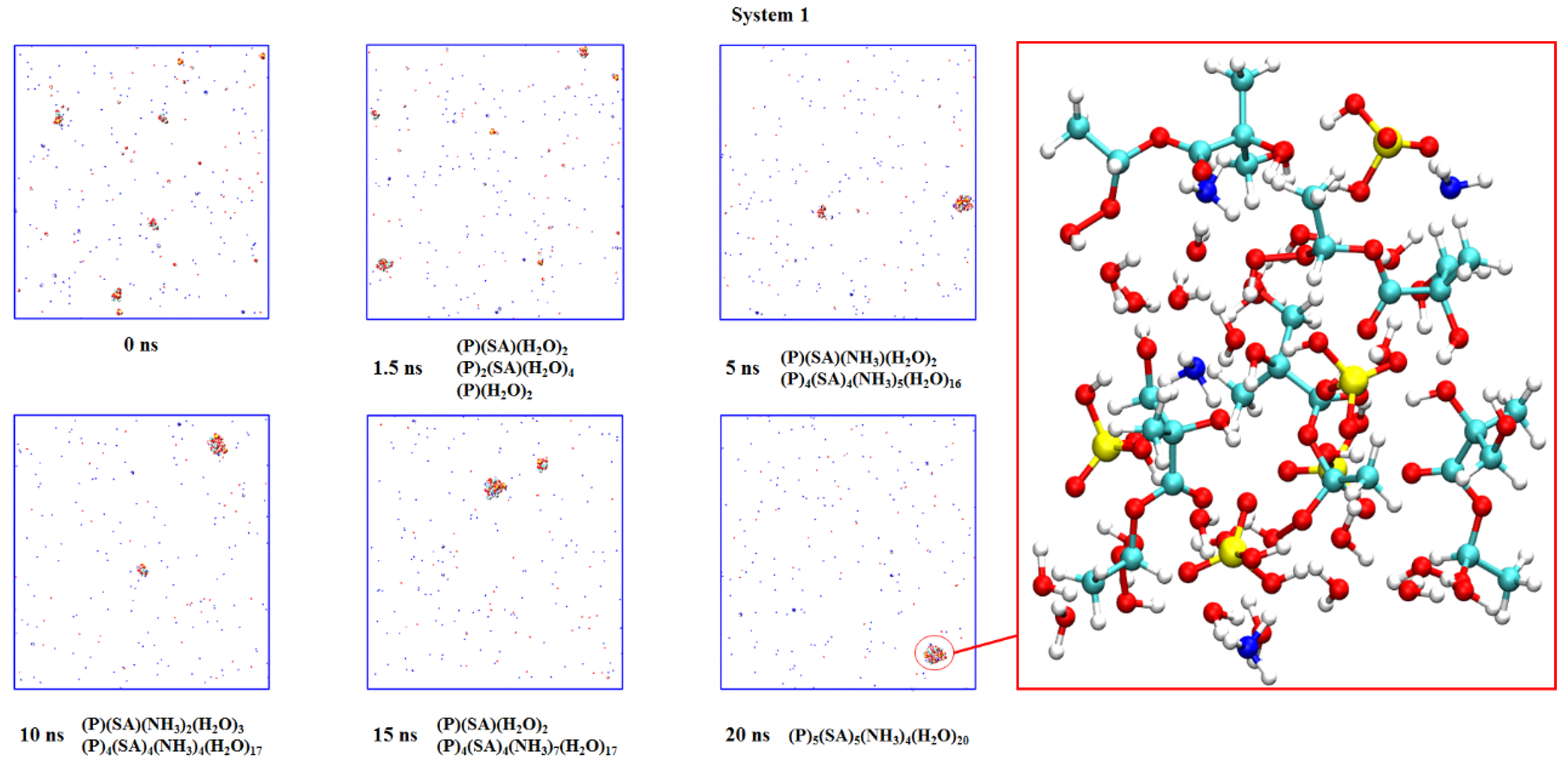
2.3. Nucleation of Products
3. Methods and Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kesselmeier, J.; Staudt, M. Biogenic volatile organic compounds (VOC): An overview on emission, physiology and ecology. J. Atmos. Chem. 1999, 33, 23–88. [Google Scholar] [CrossRef]
- Criegee, R. Mechanism of ozonolysis. Angew. Chem.-Int. Edit. Engl. 1975, 14, 745–752. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Percival, C.J.; Caravan, R.L.; Taatjes, C.A.; Shallcross, D.E. Criegee intermediates and their impacts on the troposphere. Environ. Sci. Process. Impacts 2018, 20, 437–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutbrod, R.; Schindler, R.N.; Kraka, E.; Cremer, D. Formation of OH radicals in the gas phase ozonolysis of alkenes, the unexpected role of carbonyl oxides. Chem. Phys. Lett. 1996, 252, 221–229. [Google Scholar] [CrossRef]
- Taatjes, C.A.; Shallcross, D.E.; Percival, C.J. Research frontiers in the chemistry of Criegee intermediates and tropospheric ozonolysis. Phys. Chem. Chem. Phys. 2014, 16, 1704–1718. [Google Scholar] [CrossRef]
- Emmerson, K.M.; Carslaw, N.; Carslaw, D.C.; Lee, J.D.; McFiggans, G.; Bloss, W.J.; Gravestock, T.; Heard, D.E.; Hopkins, J.; Ingham, T.; et al. Free radical modelling studies during the UK TORCH Campaign in Summer 2003. Atmos. Chem. Phys. 2007, 7, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Emmerson, K.M.; Carslaw, N. Night-time radical chemistry during the TORCH campaign. Atmos. Environ. 2009, 43, 3220–3226. [Google Scholar] [CrossRef]
- Elshorbany, Y.F.; Kurtenbach, R.; Wiesen, P.; Lissi, E.; Rubio, M.; Villena, G.; Gramsch, E.; Rickard, A.R.; Pilling, M.J.; Kleffmann, J. Oxidation capacity of the city air of Santiago, Chile. Atmos. Chem. Phys. 2009, 9, 2257–2273. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.M.; Yin, J.; Tilling, R.M.; Cai, X.; Seakins, P.W.; Hopkins, J.R.; Lansley, D.L.; Lewis, A.C.; Hunter, M.C.; Heard, D.E.; et al. Measurement and modelling of air pollution and atmospheric chemistry in the U.K. West Midlands conurbation: Overview of the PUMA Consortium project. Sci. Total Environ. 2006, 360, 5–25. [Google Scholar] [CrossRef]
- Drozd, G.T.; Donahue, N.M. Pressure dependence of stabilized Criegee intermediate formation from a sequence of alkenes. J. Phys. Chem. A 2011, 115, 4381–4387. [Google Scholar] [CrossRef]
- Tobias, H.J.; Ziemann, P.J. Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids, and water with the C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene. J. Phys. Chem. A 2001, 105, 6129–6135. [Google Scholar] [CrossRef]
- Vereecken, L.; Harder, H.; Novelli, A. The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere. Phys. Chem. Chem. Phys. 2012, 14, 14682–14695. [Google Scholar] [CrossRef]
- Ouyang, B.; McLeod, M.W.; Jones, R.L.; Bloss, W.J. NO3 radical production from the reaction between the Criegee intermediate CH2OO and NO2. Phys. Chem. Chem. Phys. 2013, 15, 17070–17075. [Google Scholar] [CrossRef]
- Chao, W.; Hsieh, J.T.; Chang, C.H.; Lin, J.J.M. Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor. Science 2015, 347, 751–754. [Google Scholar] [CrossRef]
- Osborn, D.L.; Taatjes, C.A. The physical chemistry of Criegee intermediates in the gas phase. Int. Rev. Phys. Chem. 2015, 34, 309–360. [Google Scholar] [CrossRef]
- Marklund, S. Actions of Hydroxymethylhydroperoxide and Bis(Hydroxymethyl)Peroxide on Fumarate Hydratase, Lactate-Dehydrogenase, Aspartate Aminotransferase, Glucose Oxidase, and Acid-Phosphatase. Biochim. Et Biophys. Acta 1972, 258, 9–16. [Google Scholar] [CrossRef]
- Marklund, S. Mechanisms of Irreversible Inactivation of Horseradish-Peroxidase Caused by Hydroxymethylhydroperoxide. Arch. Biochem. Biophys. 1973, 154, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Millet, D.B.; Baasandorj, M.; Farmer, D.K.; Thornton, J.A.; Baumann, K.; Brophy, P.; Chaliyakunnel, S.; de Gouw, J.A.; Graus, M.; Hu, L.; et al. A large and ubiquitous source of atmospheric formic acid. Atmos. Chem. Phys. 2015, 15, 6283–6304. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.M.; Hong, S.; Fang, W.J.; Zheng, R.H.; Qin, Y.D. Formation of OH radicals from the simplest Criegee intermediate CH2OO and water. Theor. Chem. Acc. 2019, 138, 13. [Google Scholar] [CrossRef]
- Docherty, K.S.; Ziemann, P.J. Effects of Stabilized Criegee Intermediate and OH Radical Scavengers on Aerosol Formation from Reactions of β-Pinene with O3. Aerosol Sci. Technol. 2003, 37, 877–891. [Google Scholar] [CrossRef]
- Caravan, R.L.; Khan, M.A.H.; Rotavera, B.; Papajak, E.; Antonov, I.O.; Chen, M.W.; Au, K.; Chao, W.; Osborn, D.L.; Lin, J.J.; et al. Products of Criegee intermediate reactions with NO2: Experimental measurements and tropospheric implications. Faraday Discuss. 2017, 200, 313–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percival, C.J.; Welz, O.; Eskola, A.J.; Savee, J.D.; Osborn, D.L.; Topping, D.O.; Lowe, D.; Utembe, S.R.; Bacak, A.; McFiggans, G.; et al. Regional and global impacts of Criegee intermediates on atmospheric sulphuric acid concentrations and first steps of aerosol formation. Faraday Discuss. 2013, 165, 45–73. [Google Scholar] [CrossRef]
- Onel, L.; Lade, R.; Mortiboy, J.; Blitz, M.A.; Seakins, P.W.; Heard, D.E.; Stone, D. Kinetics of the gas phase reaction of the Criegee intermediate CH2OO with SO2 as a function of temperature. Phys. Chem. Chem. Phys. 2021, 23, 19415–19423. [Google Scholar] [CrossRef] [PubMed]
- Howes, N.U.M.; Mir, Z.S.; Blitz, M.A.; Hardman, S.; Lewis, T.R.; Stone, D.; Seakins, P.W. Kinetic studies of C1 and C2 Criegee intermediates with SO2 using laser flash photolysis coupled with photoionization mass spectrometry and time resolved UV absorption spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 22218–22227. [Google Scholar] [CrossRef]
- McGillen, M.R.; Curchod, B.F.E.; Chhantyal-Pun, R.; Beames, J.M.; Watson, N.; Khan, M.A.H.; McMahon, L.; Shallcross, D.E.; Orr-Ewing, A.J. Criegee Intermediate-Alcohol Reactions, A Potential Source of Functionalized Hydroperoxides in the Atmosphere. ACS Earth Space Chem. 2017, 1, 664–672. [Google Scholar] [CrossRef]
- Lin, Y.H.; Yin, C.T.; Lin, W.H.; Li, Y.L.; Takahashi, K.; Lin, J.J.M. Criegee Intermediate Reaction with Alcohol Is Enhanced by a Single Water Molecule. J. Phys. Chem. Lett. 2018, 9, 7040–7044. [Google Scholar] [CrossRef]
- Chhantyal-Pun, R.; Rotavera, B.; McGillen, M.R.; Khan, M.A.H.; Eskola, A.J.; Caravan, R.L.; Blacker, L.; Tew, D.P.; Osborn, D.L.; Percival, C.J.; et al. Criegee Intermediate Reactions with Carboxylic Acids: A Potential Source of Secondary Organic Aerosol in the Atmosphere. ACS Earth Space Chem. 2018, 2, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Joudan, S.; Forbes, M.W.; Zhou, Z.; Abbatt, J.P.D. Reaction of Condensed-Phase Criegee Intermediates with Carboxylic Acids and Perfluoroalkyl Carboxylic Acids. Environ. Sci. Technol. Lett. 2019, 6, 243–250. [Google Scholar] [CrossRef]
- Yin, C.; Takahashi, K. Effect of unsaturated substituents in the reaction of Criegee intermediates with water vapor. Phys. Chem. Chem. Phys. 2018, 20, 20217–20227. [Google Scholar] [CrossRef]
- Cabezas, C.; Guillemin, J.C.; Endo, Y. Probing the conformational behavior of the doubly substituted methyl-ethyl Criegee intermediate by FTMW spectroscopy. J. Chem. Phys. 2017, 146, 174304. [Google Scholar] [CrossRef]
- Zhao, F.; Feng, Y.J.; Liu, Y.R.; Jiang, S.; Huang, T.; Wang, Z.H.; Xu, C.X.; Huang, W. Enhancement of Atmospheric Nucleation by Highly Oxygenated Organic Molecules: A Density Functional Theory Study. J. Phys. Chem. A 2019, 123, 5367–5377. [Google Scholar] [CrossRef]
- Zhang, H.; Surratt, J.D.; Lin, Y.H.; Bapat, J.; Kamens, R.M. Effect of relative humidity on SOA formation from isoprene/NO photooxidation: Enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions. Atmos. Chem. Phys. 2011, 11, 6411–6424. [Google Scholar] [CrossRef] [Green Version]
- Jaoui, M.; Corse, E.W.; Lewandowski, M.; Offenberg, J.H.; Kleindienst, T.E.; Edney, E.O. Formation of organic tracers for isoprene SOA under acidic conditions. Atmos. Environ. 2010, 44, 1798–1805. [Google Scholar] [CrossRef]
- Edney, E.O.; Kleindienst, T.E.; Jaoui, M.; Lewandowski, M.; Offenberg, J.H.; Wang, W.; Claeys, M. Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOX/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States. Atmos. Environ. 2005, 39, 5281–5289. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.; Xie, Z.; Zhang, Z.; Sun, L. Impacts of Siberian biomass burning on organic aerosols over the North Pacific Ocean and the Arctic: Primary and secondary organic tracers. Environ. Sci. Technol. 2013, 47, 3149–3157. [Google Scholar] [CrossRef]
- Hu, Q.H.; Xie, Z.Q.; Wang, X.M.; Kang, H.; He, Q.F.; Zhang, P. Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic. Sci. Rep. 2013, 3, 2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ion, A.C.; Vermeylen, R.; Kourtchev, I.; Cafmeyer, J.; Chi, X.; Gelencser, A.; Maenhaut, W.; Claeys, M. Polar organic compounds in rural PM2.5 aerosols from K-puszta, Hungary, during a 2003 summer field campaign: Sources and diel variations. Atmos. Chem. Phys. 2005, 5, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.B.; Laskin, J.; Laskin, A.; Nizkorodov, S.A. Nitrogen-Containing Organic Compounds and Oligomers in Secondary Organic Aerosol Formed by Photooxidation of Isoprene. Environ. Sci. Technol. 2011, 45, 6908–6918. [Google Scholar] [CrossRef] [PubMed]
- Vereecken, L. The reaction of Criegee intermediates with acids and enols. Phys. Chem. Chem. Phys. 2017, 19, 28630–28640. [Google Scholar] [CrossRef] [PubMed]
- Welz, O.; Eskola, A.J.; Sheps, L.; Rotavera, B.; Savee, J.D.; Scheer, A.M.; Osborn, D.L.; Lowe, D.; Booth, A.M.; Xiao, P.; et al. Rate Coefficients of C1 and C2 Criegee Intermediate Reactions with Formic and Acetic Acid Near the Collision Limit: Direct Kinetics Measurements and Atmospheric Implications. Angew. Chem.-Int. Edit. 2014, 53, 4547–4550. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Shi, Q.; Davidovits, P.; Worsnop, D.R.; Zahniser, M.S.; Kolb, C.E. Reactive Uptake of CI2(g) and Br2(g) by Aqueous Surfaces as a Function of Br- and I- Ion Concentration: The Effect of Chemical Reaction at the Interface. J. Phys. Chem. 1995, 99, 8768–8776. [Google Scholar] [CrossRef]
- Magi, L.; Schweitzer, F.; Pallares, C.; Cherif, S.; Mirabel, P.; George, C. Investigation of the uptake rate of ozone and methyl hydroperoxide by water surfaces. J. Phys. Chem. A 1997, 101, 4943–4949. [Google Scholar] [CrossRef]
- Gerber, R.B.; Varner, M.E.; Hammerich, A.D.; Riikonen, S.; Murdachaew, G.; Shemesh, D.; Finlayson-Pitts, B.J. Computational studies of atmospherically-relevant chemical reactions in water clusters and on liquid water and ice surfaces. Acc. Chem. Res. 2015, 48, 399–406. [Google Scholar] [CrossRef]
- Aplincourt, P.; Ruiz-Lopez, M.F. Theoretical study of formic acid anhydride formation from carbonyl oxide in the atmosphere. J. Phys. Chem. A 2000, 104, 380–388. [Google Scholar] [CrossRef]
- Atkinson, R. Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes. J. Phys. Chem. Ref. Data 1997, 26, 215–290. [Google Scholar] [CrossRef] [Green Version]
- Cabezas, C.; Endo, Y. The reactivity of the Criegee intermediate CH(3)CHOO with water probed by FTMW spectroscopy. J. Chem. Phys. 2018, 148, 014308. [Google Scholar] [CrossRef] [PubMed]
- Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ. 1996, 31, 81–104. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Y.; Xue, Y.; Cao, J.; Wang, W. Effect of oligomerization reactions of Criegee intermediate with organic acid/peroxy radical on secondary organic aerosol formation from isoprene ozonolysis. Atmos. Environ. 2018, 187, 218–229. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Y.; Xue, Y.; Jia, Z.; Wang, W. Oligomer formation from the gas-phase reactions of Criegee intermediates with hydroperoxide esters: Mechanism and kinetics. Atmos. Chem. Phys. 2022, 22, 14529–14546. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Fukui, K. The path of chemical-reactions—The IRC approach. Accounts Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 9. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef] [Green Version]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Groups | Reactant Serial Numbers | Reactants | Reaction Energy Barriers (kcal/mol) | |
|---|---|---|---|---|
| Syn-CH3CHOO | Anti-CH3CHOO | |||
| COOH | R1 |  | 0.00 | 0.00 |
| R2 |  | 0.06 | 0.00 | |
| R3 |  | 0.00 | 0.00 | |
| α-OH | R4 |  | 13.67 | 7.84 |
| R2 |  | 16.83 | 8.46 | |
| R5 |  | 13.37 | 7.99 | |
| β-OH | R4 |  | 15.30 | 9.63 |
| R1 |  | 13.67 | 7.52 | |
| R6 |  | 12.65 | 5.90 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhang, Q.; Wei, Y.; Wang, Q.; Wang, W. Theoretical Study on the Gas-Phase and Aqueous Interface Reaction Mechanism of Criegee Intermediates with 2-Methylglyceric Acid and the Nucleation of Products. Int. J. Mol. Sci. 2023, 24, 5400. https://doi.org/10.3390/ijms24065400
Li L, Zhang Q, Wei Y, Wang Q, Wang W. Theoretical Study on the Gas-Phase and Aqueous Interface Reaction Mechanism of Criegee Intermediates with 2-Methylglyceric Acid and the Nucleation of Products. International Journal of Molecular Sciences. 2023; 24(6):5400. https://doi.org/10.3390/ijms24065400
Chicago/Turabian StyleLi, Lei, Qingzhu Zhang, Yuanyuan Wei, Qiao Wang, and Wenxing Wang. 2023. "Theoretical Study on the Gas-Phase and Aqueous Interface Reaction Mechanism of Criegee Intermediates with 2-Methylglyceric Acid and the Nucleation of Products" International Journal of Molecular Sciences 24, no. 6: 5400. https://doi.org/10.3390/ijms24065400