
PCSK9 Inhibitors Reduce PCSK9 and Early Atherogenic Biomarkers in Stimulated Human Coronary Artery Endothelial Cells


Abstract
:1. Introduction
2. Results
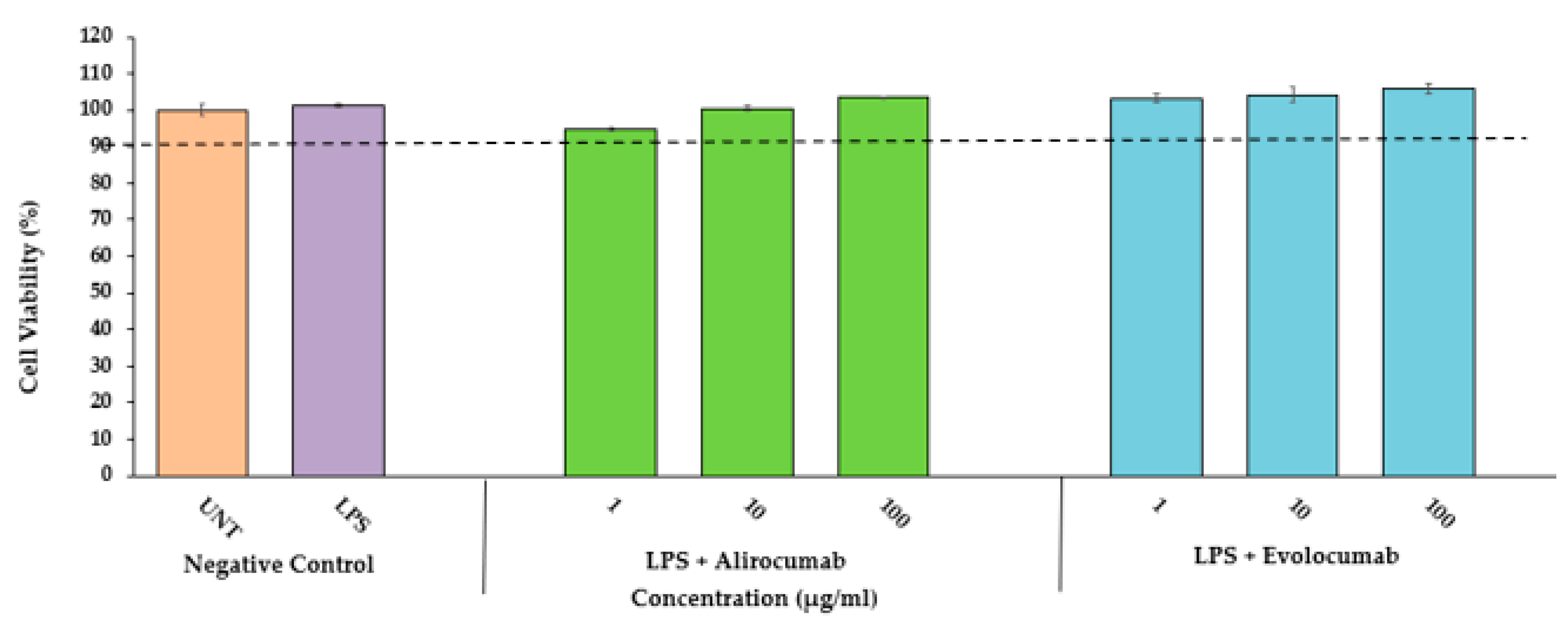
2.1. Cytotoxic Effects of PCSK9 Inhibitors on HCAEC
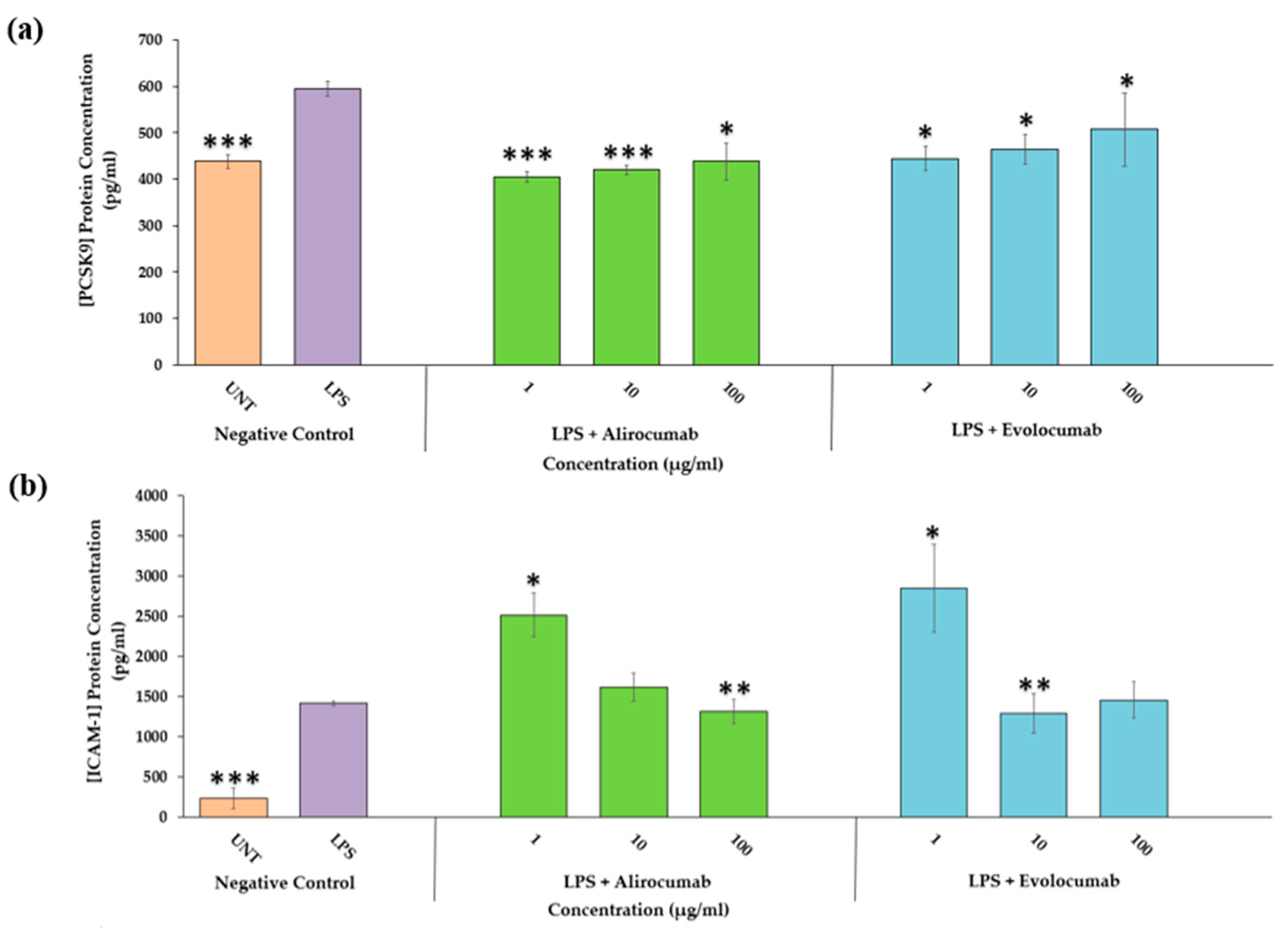
2.2. Effects of PCSK9 Inhibitors on PCSK9 Protein Expression
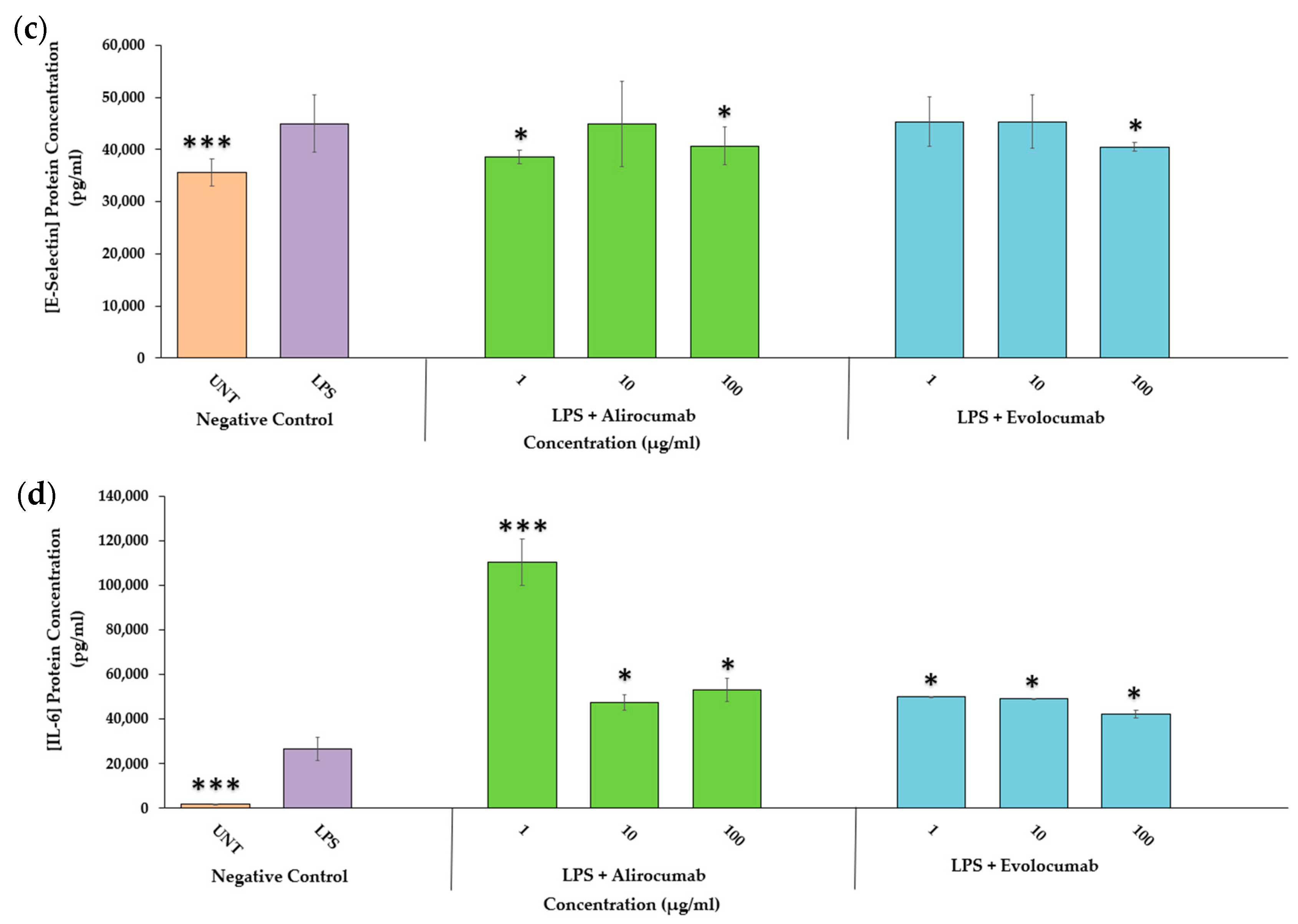
2.3. Effects of PCSK9 Inhibitors on Endothelial Activation Protein Expression (ICAM-1 and E-Selectin)
2.4. Effects of PCSK9 Inhibitors on IL-6 Protein Expression
2.5. Effects of PCSK9 Inhibitor on PCSK9 Gene Expression
2.6. Effects of PCSK9 Inhibitor on Endothelial Activation Gene Expression (ICAM-1 and E-Selectin)
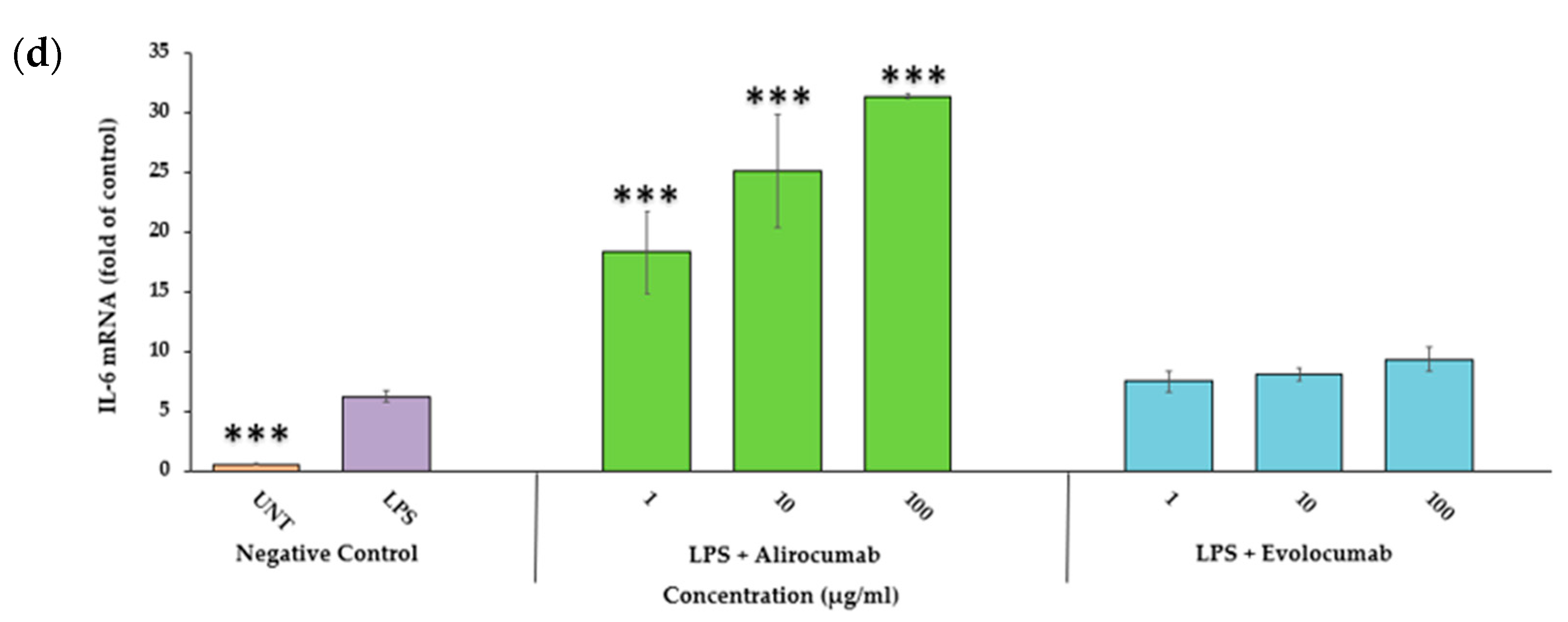
2.7. Effects of PCSK9 Inhibitor on IL-6 Gene Expression
2.8. Effects of PCSK9 Inhibitor on NF-ĸB p65 Protein and Gene Expression
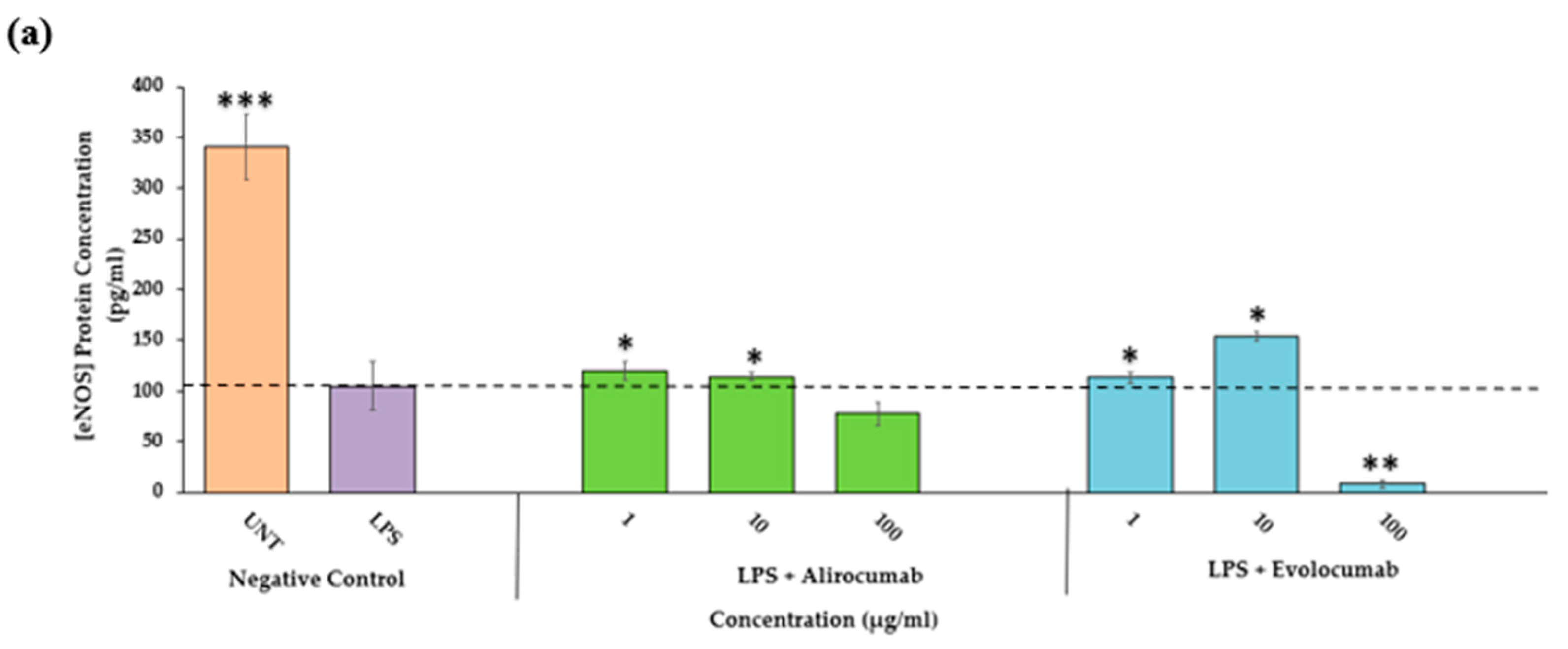
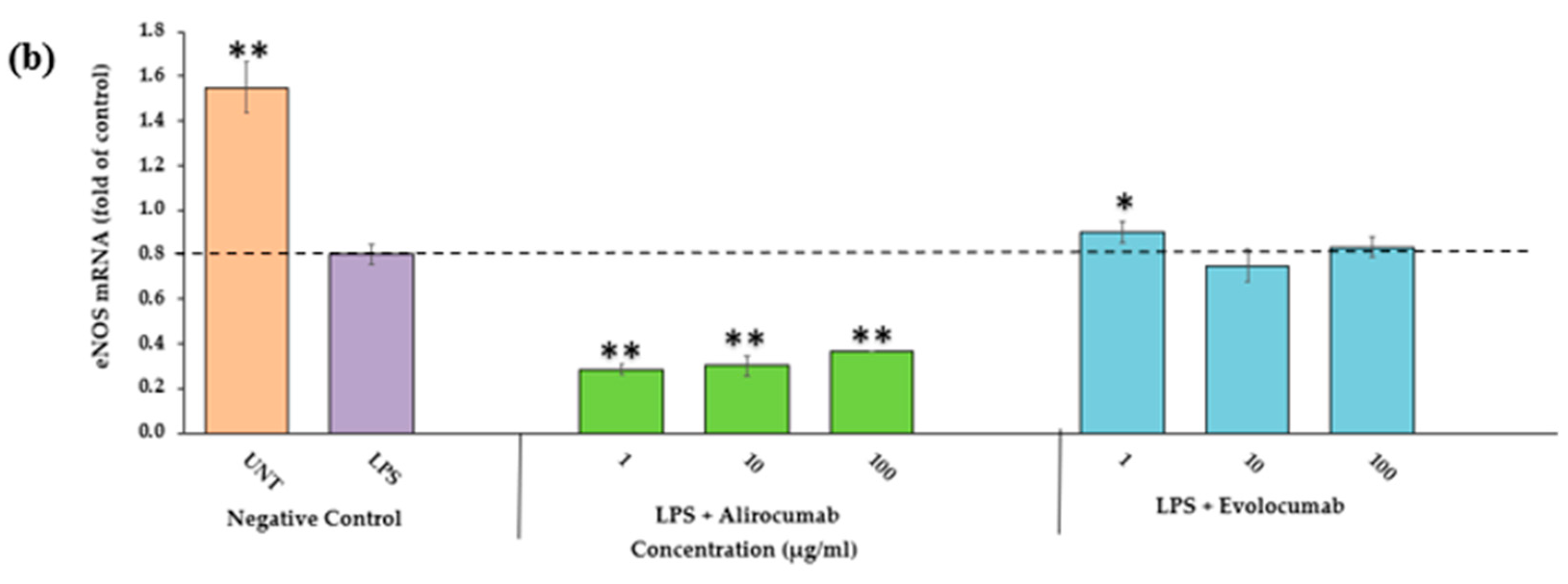
2.9. Effects of PCSK9 Inhibitor on eNOS Protein and Gene Expressions
2.10. Effects of PCSK9 Inhibitors on Monocytes and LPS-Stimulated HCAEC Interaction
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability
4.3. Treatment of HCAEC
4.4. Samples Collection for Protein Measurement
4.4.1. Cell Culture Supernatant Collection
4.4.2. Nuclear Lysates Collection (Nuclear Extraction) for NF-ĸB Assay
4.4.3. Cell Lysates Collection for eNOS Assay
4.5. Measurement of Protein Expression
4.5.1. Quantitation of PCSK9, IL-6, ICAM-1, and E-Selectin in the Supernatant
4.5.2. Measurement of NF-κB p65 Protein in Nuclear Lysates Protein
4.5.3. Quantitation of eNOS Protein in Cell Lysates
4.6. Gene Expression
4.6.1. RNA Extraction
4.6.2. Quantitation of PCSK9, Inflammation, Endothelial Activation, NF-κB, and eNOS Genes
4.6.3. Monocyte Binding Assay
4.6.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.N.; Husain, K.; Jubri, Z.; Chan, K.M.; Jantan, I.; Mohd Fauzi, N. Gynura procumbens (Lour.) Merr. extract attenuates monocyte adherence to endothelial cells through suppression of the NF-κB signaling pathway. J. Ethnopharmacol. 2022, 10, 294. [Google Scholar] [CrossRef]
- Mohd Ariff, A.; Abu Bakar, N.A.; Abd Muid, S.; Omar, E.; Ismail, N.H.; Ali, A.M.; Mohd Kasim, N.A.; Mohd Nawawi, H. Ficus deltoidea suppresses endothelial activation, inflammation, monocytes adhesion, and oxidative stress via NF-κB and eNOS pathways in stimulated human coronary artery endothelial cells. BMC Complement. Med. Ther. 2020, 17, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 1, 8563845. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.A.; Gipe, D.; Bergeron, J. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A phase 2 randomised controlled trial. Lancet 2012, 380, 29–36. [Google Scholar] [CrossRef]
- Ridker, P.M.; Revkin, J.; Amarenco, P.; Brunell, R.; Curto, M.; Civeira, F.; Flather, M.; Glynn, R.; Gregoire, J.; Jukema, J.; et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N. Engl. J. Med. 2017, 376, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- StatPearls. PCSK9 Inhibitors. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448100/#_NBK448100_pubdet_ (accessed on 31 December 2022).
- Roth, E.M.; McKenney, J.M.; Hanotin, C.; Asset, G.; Stein, E.A. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N. Engl. J. Med. 2012, 367, 1891–1900. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tecson, K.M.; Rocha, N.A.; McCullough, P.A. Usefulness of alirocumab and evolocumab for the treatment of patients with diabetic dyslipidemia. In Baylor University Medical Center Proceedings; Taylor & Francis: Abingdon, UK, 2018; Volume 11, pp. 180–184. [Google Scholar] [CrossRef] [Green Version]
- Dixon, D.L.; Pamulapati, L.G.; Bucheit, J.D.; Sisson, E.M.; Smith, S.R.; Kim, C.J.; Wohlford, G.F.; Pozen, J. Recent Updates on the Use of PCSK9 Inhibitors in Patients with Atherosclerotic Cardiovascular Disease. Curr. Atheroscler. Rep. 2019, 16, 16. [Google Scholar] [CrossRef]
- Dong, B.; Wu, M.; Li, H.; Kraemer, F.B.; Adeli, K.; Seidah, N.G.; Park, S.W.; Liu, J. Strong induction of PCSK9 gene expression through HNF1α and SREBP2: Mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid. Res. 2010, 51, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Li, S.L.; Hu, J.H.; Sun, K.J.; Liu, L.L.; Xu, D.Y. Research progress on alternative non-classical mechanisms of PCSK9 in atherosclerosis in patients with and without diabetes. Cardiovasc. Diabetol. 2020, 19, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zulkapli, R.; Yusof, M.Y.P.M.; Abd Muid, S.; Wang, S.M.; Firus Khan, A.Y.; Nawawi, H.A. Systematic Review on Attenuation of PCSK9 in Relation to Atherogenesis Biomarkers Associated with Natural Products or Plant Bioactive Compounds in In Vitro Studies: A Critique on the Quality and Imprecision of Studies. Int. J. Environ. Res. Public Health 2022, 19, 12878. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.R.; Kohli, P.; Giugliano, R.P.; O’Donoghue, M.L.; Somaratne, R.; Zhou, J.; Hoffman, E.B.; Huang, F.; Rogers, W.J.; Wasserman, S.M.; et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: An analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation 2013, 128, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Massimiliano, R.; Maria, F.G.; Nicola, F.; Alberto, C. Lipoprotein(a) and PCSK9 inhibition: Clinical evidence. Eur. Heart J. 2020, 22, 53–56. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Desai, N.R.; Kohli, P.; Rogers, W.J.; Somaratne, R.; Huang, F.; Liu, T.; Mohanavelu, S.; Hoffman, E.B.; McDonald, S.T.; et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): A randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012, 380, 2007–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, D.J.; Hala, T.; Bolognese, M.; Lillestol, M.J.; Toth, P.D.; Burges, L.; Ceska, R.; Roth, E.; Koren, M.J.; Ballantyne, C.M.; et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N. Engl. J. Med. 2014, 370, 1809–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, M.J.; Lundqvist, P.; Bolognese, M.; Neutel, J.M.; Monsalvo, M.L.; Yang, J.; Kim, J.B.; Scott, R.; Wasserman, S.M.; Bays, H. Anti-PCSK9 monotherapy for hypercholesterolemia: The MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2531–2540. [Google Scholar] [CrossRef] [Green Version]
- Koren, M.J.; Scott, R.; Kim, J.B.; Knusel, B.; Liu, T.; Lei, L.; Bolognese, M.; Wasserman, S.M. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): A randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012, 380, 1995–2006. [Google Scholar] [CrossRef]
- Raal, F.; Scott, R.; Somaratne, R.; Bridges, I.; Li, G.; Wasserman, S.M.; Stein, E.A. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: The reduction of LDL-C with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder (RUTHERFORD) randomized trial. Circulation 2012, 126, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. PCSK9 inhibitors: A new era of lipid lowering therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.; Honarpour, N.; Wang, H.; Liu, T.; Wasserman, S.M.; Scott, R.; Sever, P.S.; Pedersen, T.R. Rationale and design of the Further cardiovascular Outcomes Research with PCSK9 Inhibition in subjects with Elevated Risk trial. Am. Heart J. 2016, 173, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Keech, A.C.; Oyama, K.; Sever, P.S.; Tang, M.; Murphy, S.A.; Hirayama, A.; Lu, C.; Tay, L.; Deedwania, P.C.; Siu, C.W.; et al. Efficacy and Safety of Long-Term Evolocumab Use Among Asian Subjects-A Subgroup Analysis of the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER). Trial. Circ. J. 2021, 85, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.A.; Pedersen, T.R.; Gaciong, Z.A.; Ceska, R.; Ezhov, M.V.; Connolly, D.L.; Jukema, J.W.; Toth, K.; Tikkanen, M.J.; Im, K.; et al. Effect of the PCSK9 Inhibitor Evolocumab on Total Cardiovascular Events in Patients with Cardiovascular Disease: A Prespecified Analysis from the FOURIER Trial. JAMA Cardiol. 2019, 4, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Sinnaeve, P.R.; Schwartz, G.G.; Wojdyla, D.M.; Alings, M.; Bhatt, D.L.; Bittner, V.A.; Chiang, C.E.; Correa Flores, R.M.; Diaz, R.; Dorobantu, M.; et al. Effect of alirocumab on cardiovascular outcomes after acute coronary syndromes according to age: An ODYSSEY OUTCOMES trial analysis. Eur. Heart J. 2020, 21, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, E.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Danchin, N.; Diaz, R.; Goodman, S.G.; Harrington, R.A.; Jukema, J.W.; et al. Apolipoprotein B, Residual Cardiovascular Risk After Acute Coronary Syndrome, and Effects of Alirocumab. Circulation. 2022, 146, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; McGinniss, J.; Steg, P.G.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Dilic, M.; Goodman, S.G.; Jukema, J.W.; Loy, M.; et al. Achievement of ESC/EAS LDL-C treatment goals after an acute coronary syndrome with statin and alirocumab. Eur. J. Prev. Cardiol. 2022, 29, 1842–1851. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J. Effect of evolocumab on progression of coronary disease in statin-treated patients: The GLAGOV randomised clinical trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Karagiannis, A.D.; Liu, M.; Toth, P.P.; Zhao, S.; Agrawal, D.K.; Libby, P.; Chatzizisis, Y.S. Pleiotropic Anti-atherosclerotic Effects of PCSK9 Inhibitors from Molecular Biology to Clinical Translation. Curr. Atheroscler. Rep. 2018, 10, 20. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Fazio, S. PCSK9 and atherosclerosis-lipids and beyond. J. Atheroscler. Thromb. 2017, 24, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK9. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luquero, A.; Badimon, L.; Borrell-Pages, M. PCSK9 Functions in Atherosclerosis Are Not Limited to Plasmatic LDL-Cholesterol Regulation. Front. Cardiovasc. Med. 2021, 23, 639727. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, R.; Basta, G.; Neglia, D.; De Caterina, R.; Del Turco, S.; Caselli, C. PCSK9 and atherosclerosis: Looking beyond LDL regulation. Eur. J. Clin. Investig. 2021, 51, e13459. [Google Scholar] [CrossRef] [PubMed]
- Tromp, T.R.; Hartgers, M.L.; Hovingh, G.K.; Vallejo-Vaz, A.J.; Ray, K.K.; Soran, H.; Freiberger, T.; Bertolini, S.; Harada-Shiba, M.; Blom, D.J.; et al. Homozygous Familial Hypercholesterolaemia International Clinical Collaborators. Worldwide experience of homozygous familial hypercholesterolaemia: Retrospective cohort study. Lancet. 2022, 19, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Sizar, O.; Khare, S.; Jamil, R.T.; Talati, R. Statin Medications; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430940/ (accessed on 31 December 2022).
- Al-Khateeb, A.; Zahri, M.K.; Mohamed, M.S.; Sasongko, T.H.; Ibrahim, S.; Yusof, Z.; Zilfalil, B.A. Analysis of sequence variations in low-density lipoprotein receptor gene among Malaysian patients with familial hypercholesterolemia. BMC Med. Genet. 2011, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Safaeian, L.; Vaseghi, G.; Jabari, H.; Dana, N. Evolocumab, a proprotein convertase subtilisin/kexin type 9 inhibitor, promotes angiogenesis in vitro. Can. J. Physiol. Pharmacol. 2019, 97, 352–358. [Google Scholar] [CrossRef]
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H2O2-induced oxidative stress. Arch. Physiol. Biochem. 2020, 3, 1–6. [Google Scholar] [CrossRef]
- 42. Lebeau, P.F.; Byun, J.H.; Platko, K.; Al-Hashimi, A.A.; Lhotak, S.; MacDonald, M.E.; Mejia-Benitez, A.; Prat, A.; Igdoura, S.A.; Trigatti, B. PCSK9 knockout exacerbates diet-induced non-alcoholic steatohepatitis, fibrosis and liver injury in mice. JHEP Rep. 2019, 1, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, A.; Gentile, M.; Iannuzzo, G.; Calcaterra, I.; Tripaldella, M.; Porro, B.; Cavalca, V.; Di Taranto, M.D.; Tremoli, E.; Fortunato, G.; et al. Endothelial function improvement in patients with familial hypercholesterolemia receiving PCSK-9 inhibitors on top of maximally tolerated lipid lowering therapy. Thromb. Res. 2020, 194, 229–236. [Google Scholar] [CrossRef]
- Maulucci, G.; Cipriani, F.; Russo, D.; Casavecchia, G.; Di Staso, C.; Di Martino, L.F.M.; Ruggiero, A.; Di Biase, M.; Brunetti, N.D. Improved endothelial function after short-term therapy with evolocumab. J. Clin. Lipidol. 2018, 12, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Tibolla, G.; Pirillo, A.; Cipollone, F.; Mezzetti, A.; Pacia, S.; Corsini, A.; Catapano, A.L. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 2012, 220, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.-T.; Li, T.-H.; Wang, Z.; Wei, D.-H.; Liu, L.-S.; Zheng, X.-L.; et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Schlüter, K.D.; Wolf, A.; Schreckenberg, R. Coming Back to Physiology: Extra Hepatic Functions of Proprotein Convertase Subtilisin/Kexin Type 9. Front. Physiol. 2020, 11, 598649. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Sun, C.; Wang, Y.; Mehta, J.L. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid. Redox. Signal. 2015, 22, 760–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.S.; Chen, W.; Liu, J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, J.; Chen, L.; Wu, Q.; Yu, C. Chitosan oligosaccharides enhance lipid droplets via down-regulation of PCSK9 gene expression in HepG2 cells. Exp. Cell. Res. 2018, 366, 152–160. [Google Scholar] [CrossRef]
- Ilse, T.; Anna, E.; Daniel, J.K.; Jaap, D.; van, B. Chapter Five—Leukocytes Crossing the Endothelium: A Matter of Communication. Int. Rev. Cell Mol. Biol. 2016, 322, 281–329. [Google Scholar] [CrossRef]
- Hadad, N.; Tuval, L.; Elgazar-Carmom, V.; Levy, R.; Levy, R. Endothelial ICAM-1 protein induction is regulated by cytosolic phospholipase A2α via both NFκB and CREB transcription factors. J. Immunol. 2011, 186, 1816–1827. [Google Scholar] [CrossRef] [Green Version]
- Sawa, Y.; Tsuruga, E. The expression of E-selectin and chemokines in the cultured human lymphatic endothelium with lipopolysaccharides. J. Anat. 2008, 212, 654–663. [Google Scholar] [CrossRef]
- Shyu, A.B.; Wilkinson, M.F.; van Hoof, A. Messenger RNA regulation: To translate or to degrade. EMBO J. 2008, 27, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, S.D.; Yang, L.; Luscinskas, F.W. Endothelial ICAM-1 functions in adhesion and signaling during leukocyte recruitment. In Adhesion Molecules: Function and Inhibition. Progress in Inflammation Research; Ley, K., Ed.; Birkhauser: Basel, Switzerland, 2007; pp. 99–116. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-κB—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leucker, T.M.; Gerstenblith, G.; Schar, M.; Brown, T.T.; Jones, S.R.; Afework, Y.; Weiss, R.G.; Hays, A.G. Evolocumab, a PCSK9-Monoclonal Antibody, Rapidly Reverses Coronary Artery Endothelial Dysfunction in People Living with HIV and People with Dyslipidemia. J. Am. Heart Assoc. 2020, 9, e016263. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A redox biology and medicine concept. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Sangle, G.V.; Shen, G.X. Signaling mechanisms for oxidized LDL-induced oxidative stress and the upregulation of plasminogen activator inhibitor-I in vascular cells. Clin. Lipidol. 2010, 5, 221–232. [Google Scholar] [CrossRef]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti PCSK 9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS 2 randomized, placebo controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, P.M.; Jacobson, T.A.; Bruckert, E.; Thompson, P.D.; Guyton, J.R.; Baccara-Dinet, M.T.; Gipe, D.I. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK-9, in statin-intolerant patients: Design and rationale of ODYSSEY ALTERNATIVE, a randomized phase 3 trial. J. Clin. Lipidol. 2014, 8, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, J.R.; Vadas, M.A. Endothelial adhesiveness for blood neutrophils is inhibited by transforming growth factor-beta. Science 1988, 242, 97–99. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCSK9 % | ICAM-1 % | E-Selectin % | IL-6 % | NF-κB p65 % | eNOS % | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inhibition | Inhibition | Inhibition | Inhibition | Inhibition | Increment | |||||||
| P | G | P | G | P | G | P | G | P | G | P | G | |
| Evolocumab | 5.7 | −7.5 | −3.3 | 57.4 | 3.7 | 126.9 | −132.7 | −156.7 | 21.6 | −3.8 | 65.43 | −18.6 |
| Alirocumab | 14.0 | −129.4 | −5.4 | −791.5 | 11.9 | −6761.7 | −119.2 | −1373.1 | 16.0 | −103.1 | 16.24 | −63.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zulkapli, R.; Muid, S.A.; Wang, S.M.; Nawawi, H. PCSK9 Inhibitors Reduce PCSK9 and Early Atherogenic Biomarkers in Stimulated Human Coronary Artery Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 5098. https://doi.org/10.3390/ijms24065098
Zulkapli R, Muid SA, Wang SM, Nawawi H. PCSK9 Inhibitors Reduce PCSK9 and Early Atherogenic Biomarkers in Stimulated Human Coronary Artery Endothelial Cells. International Journal of Molecular Sciences. 2023; 24(6):5098. https://doi.org/10.3390/ijms24065098
Chicago/Turabian StyleZulkapli, Rahayu, Suhaila Abd Muid, Seok Mui Wang, and Hapizah Nawawi. 2023. "PCSK9 Inhibitors Reduce PCSK9 and Early Atherogenic Biomarkers in Stimulated Human Coronary Artery Endothelial Cells" International Journal of Molecular Sciences 24, no. 6: 5098. https://doi.org/10.3390/ijms24065098