
Endoplasmic Reticulum Stress in Renal Cell Carcinoma
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Kidney and Renal Cell Carcinoma
1.1. Renal Cell Carcinoma Epidemiology and Mutational Profiles
1.2. Metabolic Reprogramming in Renal Cell Carcinoma
1.3. Therapeutic Options for Renal Cell Carcinoma
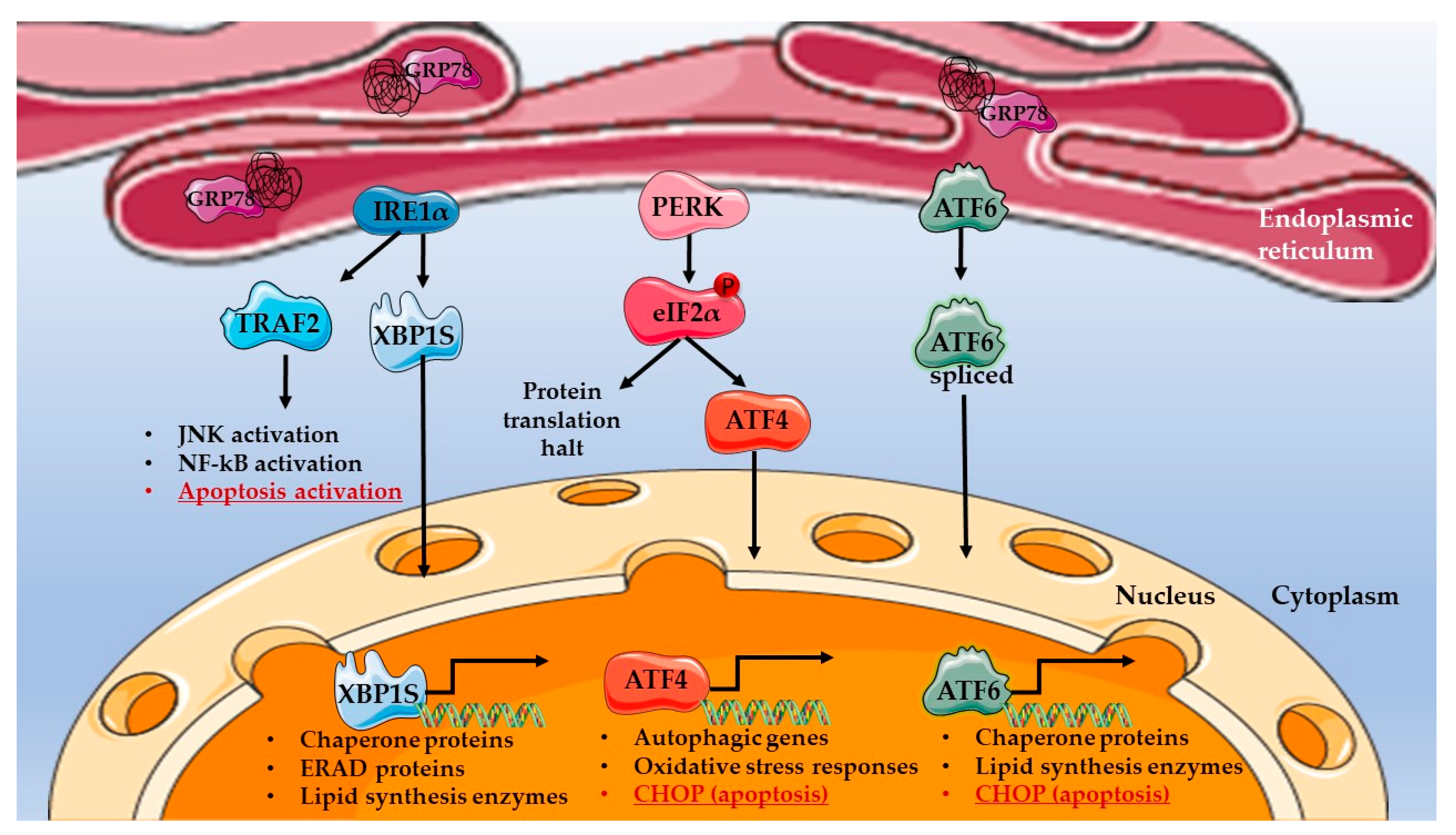
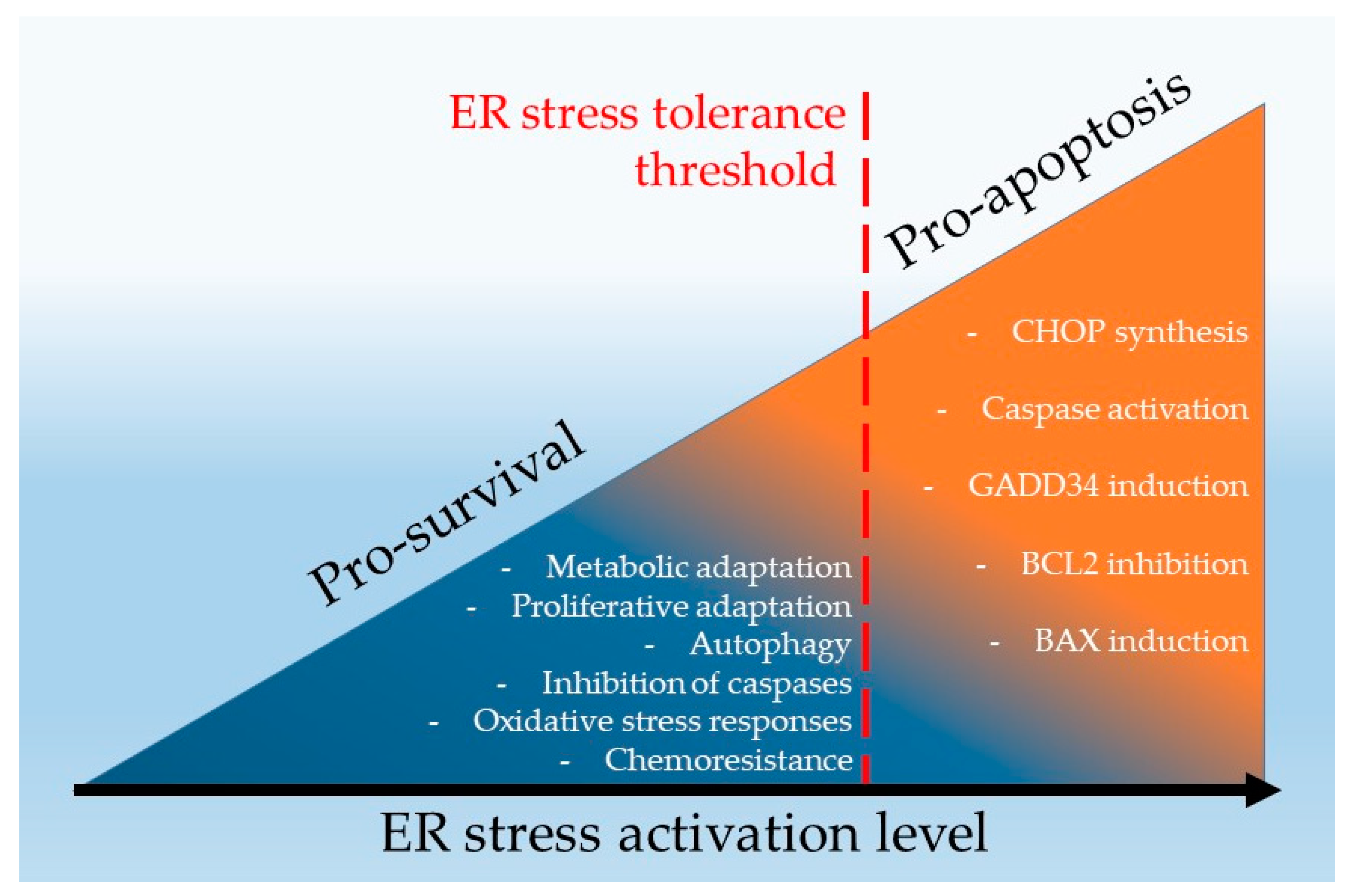
2. Endoplasmic Reticulum Stress Signalling in Pro-Survival and Pro-Apoptotic Mechanisms
3. ER Stress Signaling in Renal Cell Carcinoma
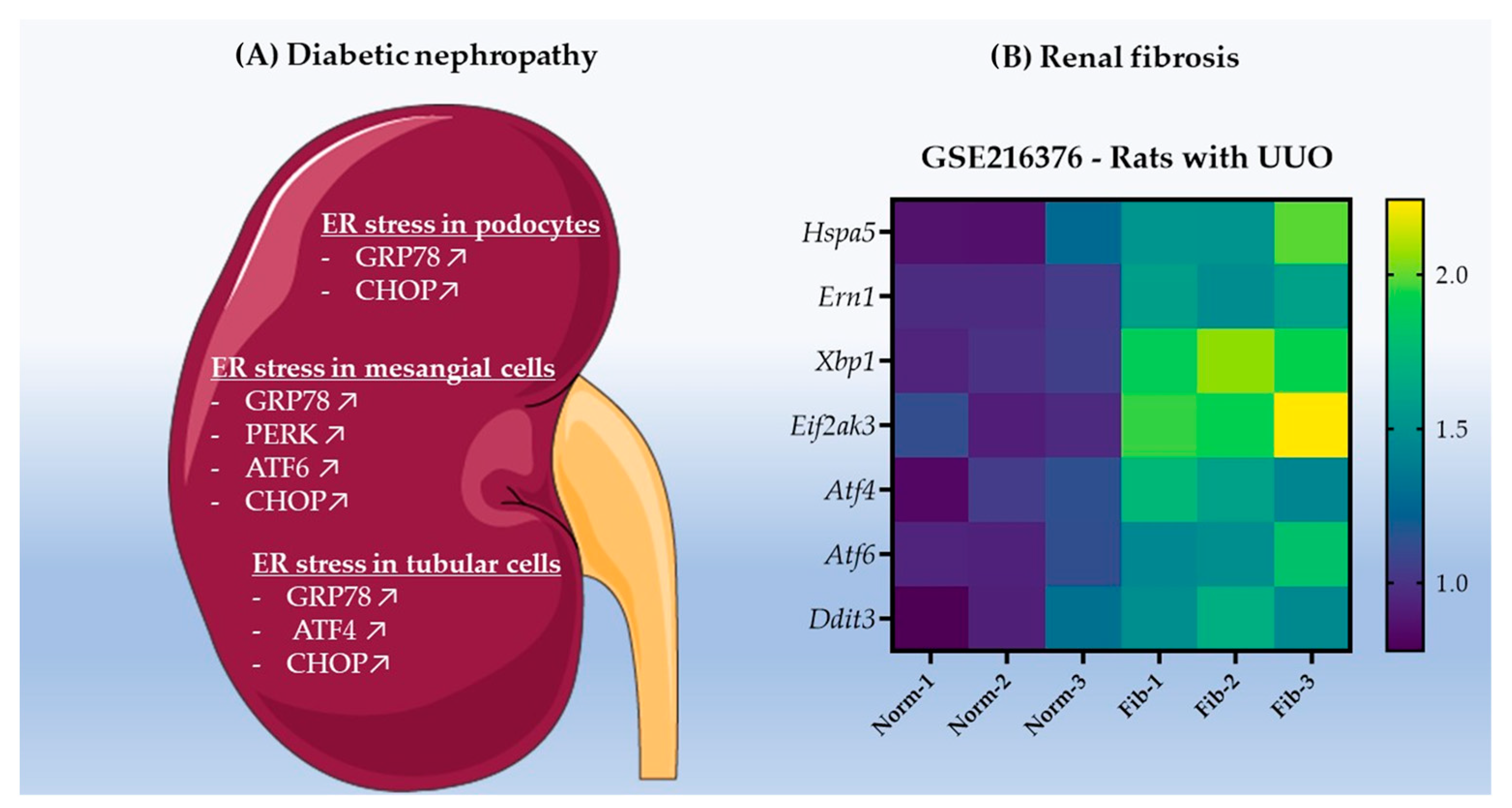
3.1. ER Stress Promotes Renal Cell Carcinoma Development in Chronic Kidney Disease
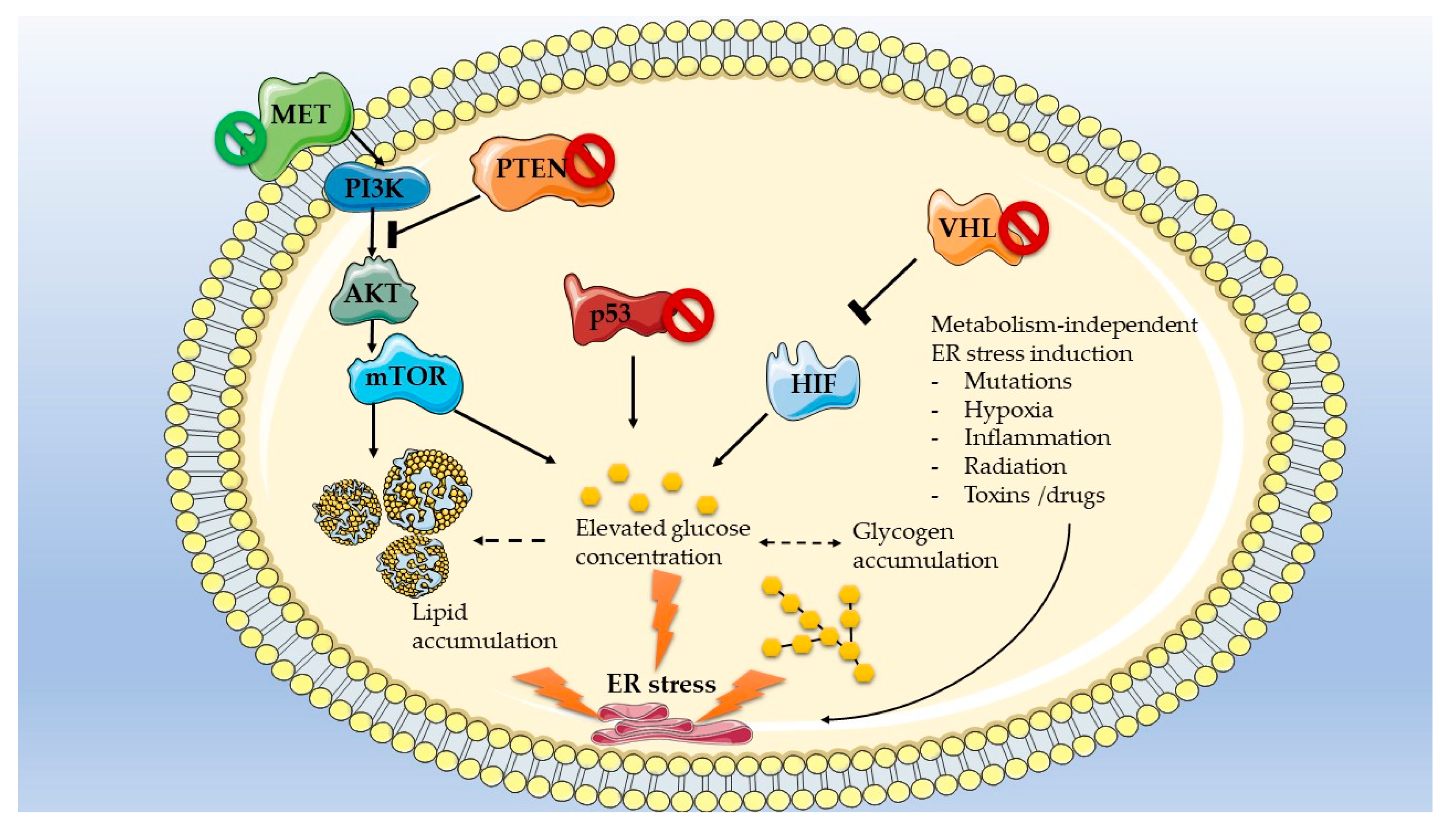
3.2. Mutations Dictate Metabolic-Dependent Activation of ER Stress in Renal Cell Carcinoma
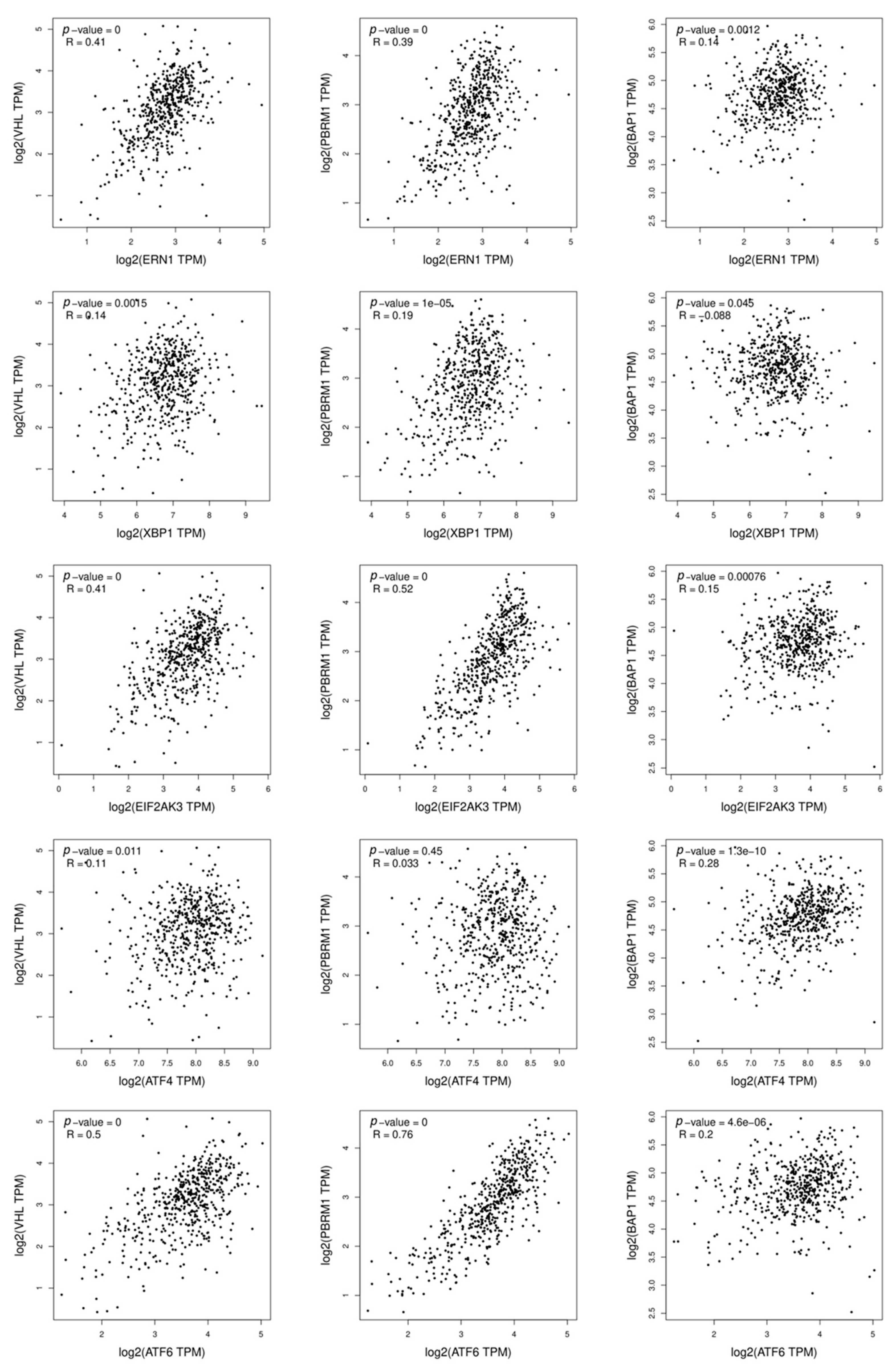
3.3. UPR Gene Mutations in Renal Cell Carcinoma
4. Targeting Endoplasmic Reticulum Stress Pathways in Renal Cancer
4.1. Pharmacological Hyperactivation of ER Stress as a Strategy to Overcome the ERST and to Induce RCC Cell Death
4.2. Nanoparticle-Mediated Hyperactivation of ER Stress in RCC
4.3. Inhibition of the UPR in Conjunction with TKI as a Therapy for RCC
4.4. Experimental Models to Investigate ER Stress in Renal Cell Carcinoma
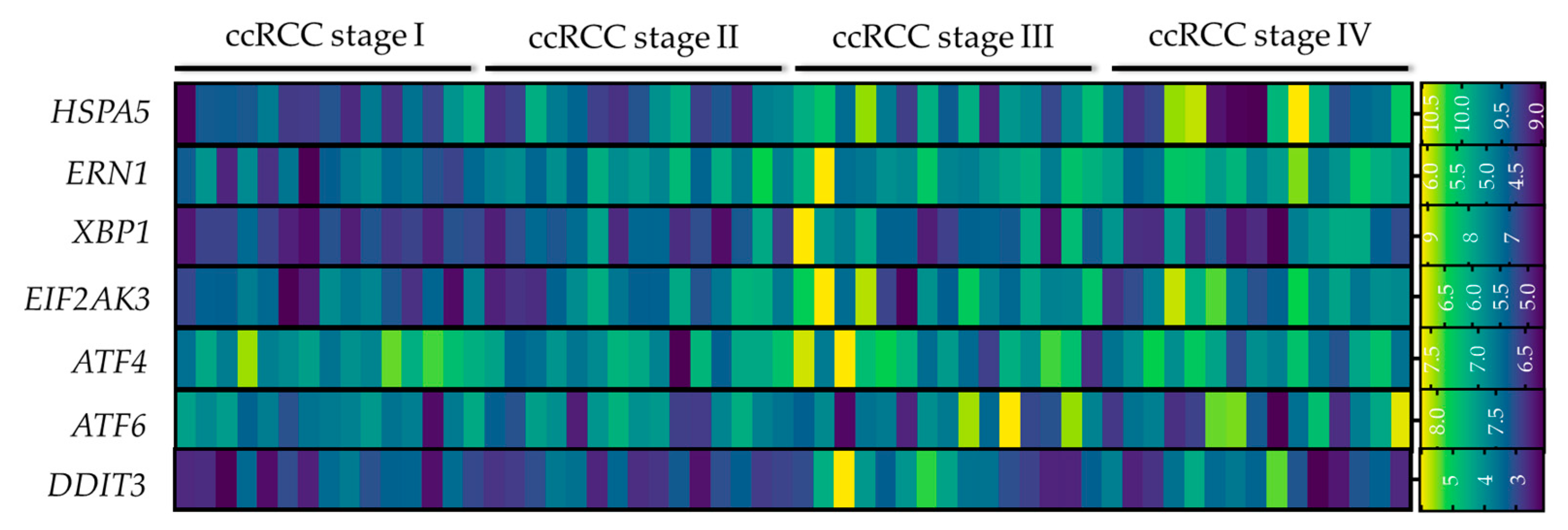
5. ER Stress as a Biomarker for RCC Prognosis
5.1. GRP78
5.2. ER Stress Gene Signature
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balzer, M.S.; Rohacs, T.; Susztak, K. How Many Cell Types Are in the Kidney and What Do They Do? Annu. Rev. Physiol. 2022, 84, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Kidney tubules: Intertubular, vascular, and glomerular cross-talk. Curr. Opin. Nephrol. Hypertens. 2016, 25, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, I.H.; Utzschneider, K.M. The kidney’s role in systemic metabolism—Still much to learn. Nephrol. Dial. Transplant. 2017, 32, 588–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewin, L.S. Sugar or Fat? Renal Tubular Metabolism Reviewed in Health and Disease. Nutrients 2021, 13, 1580. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primer 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef]
- Maher, E.R. Hereditary renal cell carcinoma syndromes: Diagnosis, surveillance and management. World J. Urol. 2018, 36, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Li, Q.; Che, X.; Wang, Q.; Wu, G. The Uniqueness of Clear Cell Renal Cell Carcinoma: Summary of the Process and Abnormality of Glucose Metabolism and Lipid Metabolism in ccRCC. Front. Oncol. 2021, 11, 727778. [Google Scholar] [CrossRef]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Zhang, B.; Fu, Y.; Zhao, X.; Zhang, J.; Zuo, K.; Xing, Y.; Jiang, S.; Qin, Z.; et al. Single-cell chromatin accessibility landscape in kidney identifies additional cell-of-origin in heterogenous papillary renal cell carcinoma. Nat. Commun. 2022, 13, 31. [Google Scholar] [CrossRef]
- Rathmell, K.W.; Chen, F.; Creighton, C.J. Genomics of chromophobe renal cell carcinoma: Implications from a rare tumor for pan-cancer studies. Oncoscience 2015, 2, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Clark, P.E. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009, 76, 939–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büscheck, F.; Fraune, C.; Simon, R.; Kluth, M.; Hube-Magg, C.; Möller-Koop, C.; Sarper, I.; Ketterer, K.; Henke, T.; Eichelberg, C.; et al. Prevalence and clinical significance of VHL mutations and 3p25 deletions in renal tumor subtypes. Oncotarget 2020, 11, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, B.J.; Hakimi, A.A. Molecular profiling of renal cell carcinoma: Building a bridge towards clinical impact. Curr. Opin. Urol. 2016, 26, 383–387. [Google Scholar] [CrossRef]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef] [PubMed]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- Suganuma, N.; Segade, F.; Matsuzu, K.; Bowden, D.W. Differential expression of facilitative glucose transporters in normal and tumour kidney tissues. BJU Int. 2007, 99, 1143–1149. [Google Scholar] [CrossRef]
- Ambrosetti, D.; Dufies, M.; Dadone, B.; Durand, M.; Borchiellini, D.; Amiel, J.; Pouyssegur, J.; Rioux-Leclercq, N.; Pages, G.; Burel-Vandenbos, F.; et al. The two glycolytic markers GLUT1 and MCT1 correlate with tumor grade and survival in clear-cell renal cell carcinoma. PLoS ONE 2018, 13, e0193477. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Song, J.; Godfrey, J.; Riscal, R.; Skuli, N.; Nissim, I.; Simon, M.C. Glycogen metabolism is dispensable for tumour progression in clear cell renal cell carcinoma. Nat. Metab. 2021, 3, 327–336. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Büttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlösser, H.; Hartmann, A.; et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8+ T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The glucose and lipid metabolism reprogramming is grade-dependent in clear cell renal cell carcinoma primary cultures and is targetable to modulate cell viability and proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Rutigliano, M.; Sanguedolce, F.; Galleggiante, V.; Giglio, A.; Cagiano, S.; Bufo, P.; Maiorano, E.; Ribatti, D.; Ranieri, E.; et al. Increased Expression of the Autocrine Motility Factor is Associated With Poor Prognosis in Patients With Clear Cell-Renal Cell Carcinoma. Medicine 2015, 94, e2117. [Google Scholar] [CrossRef]
- Lv, Z.; Qi, L.; Hu, X.; Mo, M.; Jiang, H.; Li, Y. Identification of a Novel Glycolysis-Related Gene Signature Correlates with the Prognosis and Therapeutic Responses in Patients With Clear Cell Renal Cell Carcinoma. Front. Oncol. 2021, 11, 633950. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-Related Genes Serve as Potential Prognostic Biomarkers in Clear Cell Renal Cell Carcinoma. Oxid. Med. Cell. Longev. 2021, 2021, e6699808. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Loizzo, D.; di Meo, N.A.; Lasorsa, F.; Mastropasqua, M.; Maiorano, E.; Bizzoca, C.; Vincenti, L.; Battaglia, M.; et al. MUC1 Tissue Expression and Its Soluble Form CA15-3 Identify a Clear Cell Renal Cell Carcinoma with Distinct Metabolic Profile and Poor Clinical Outcome. Int. J. Mol. Sci. 2022, 23, 13968. [Google Scholar] [CrossRef]
- Feng, C.; Li, Y.; Li, K.; Lyu, Y.; Zhu, W.; Jiang, H.; Wen, H. PFKFB4 is overexpressed in clear-cell renal cell carcinoma promoting pentose phosphate pathway that mediates Sunitinib resistance. J. Exp. Clin. Cancer Res. 2021, 40, 308. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Arai, E.; Maekawa, K.; Ishikawa, M.; Fujimoto, H.; Taguchi, R.; Matsumoto, K.; Kanai, Y.; Saito, Y. Lipidomic Signatures and Associated Transcriptomic Profiles of Clear Cell Renal Cell Carcinoma. Sci. Rep. 2016, 6, 28932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Mijn, J.C.; Fu, L.; Khani, F.; Zhang, T.; Molina, A.M.; Barbieri, C.E.; Chen, Q.; Gross, S.S.; Gudas, L.J.; Nanus, D.M. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple HIF1α-Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2020, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.A.; Rini, B.I. Recent progress in the management of advanced renal cell carcinoma. CA Cancer J. Clin. 2007, 57, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, A.Z.; Liu, L.C.; Gupta, S.; Logan, T.F.; Singer, E.A.; Joshi, M.; Zakharia, Y.N.; Lang, J.M.; Schwarz, J.K.; Al-Janadi, A.; et al. Phase Ib/II Clinical Trial of Pembrolizumab With Bevacizumab for Metastatic Renal Cell Carcinoma: BTCRC-GU14-003. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1138–1145. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor Microenvironment Dynamics in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Tamma, R.; Rutigliano, M.; Lucarelli, G.; Annese, T.; Ruggieri, S.; Cascardi, E.; Napoli, A.; Battaglia, M.; Ribatti, D. Microvascular density, macrophages, and mast cells in human clear cell renal carcinoma with and without bevacizumab treatment. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 355.e11–355.e19. [Google Scholar] [CrossRef]
- Netti, G.S.; Lucarelli, G.; Spadaccino, F.; Castellano, G.; Gigante, M.; Divella, C.; Rocchetti, M.T.; Rascio, F.; Mancini, V.; Stallone, G.; et al. PTX3 modulates the immunoflogosis in tumor microenvironment and is a prognostic factor for patients with clear cell renal cell carcinoma. Aging 2020, 12, 7585. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, X.-D.; Zhu, G.-Q.; Li, N.; Zhou, C.-W.; Yang, C.; Zeng, M.-S. Crosstalk Between Metabolism and Immune Activity Reveals Four Subtypes With Therapeutic Implications in Clear Cell Renal Cell Carcinoma. Front. Immunol. 2022, 13, 861328. [Google Scholar] [CrossRef] [PubMed]
- Ghini, V.; Laera, L.; Fantechi, B.; Monte, F.D.; Benelli, M.; McCartney, A.; Leonardo, T.; Luchinat, C.; Pozzessere, D. Metabolomics to Assess Response to Immune Checkpoint Inhibitors in Patients with Non-Small-Cell Lung Cancer. Cancers 2020, 12, 3574. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Chiellino, S. ASSURE vs. S-TRAC: Conflicting results of adjuvant treatments for kidney cancer in the era of targeted agents and genomics. Ann. Transl. Med. 2016, 4, S14. [Google Scholar] [CrossRef] [Green Version]
- Vaishampayan, U.N. The Role of Nephrectomy for Kidney Cancer in the Era of Targeted and Immune Therapies. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2016, 35, e16–e20. [Google Scholar] [CrossRef] [PubMed]
- Dengina, N.; Tsimafeyeu, I.; Mitin, T. Current Role of Radiotherapy for Renal-Cell Carcinoma: Review. Clin. Genitourin. Cancer 2017, 15, 183–187. [Google Scholar] [CrossRef]
- Duran, I.; Lambea, J.; Maroto, P.; González-Larriba, J.L.; Flores, L.; Granados-Principal, S.; Graupera, M.; Sáez, B.; Vivancos, A.; Casanovas, O. Resistance to Targeted Therapies in Renal Cancer: The Importance of Changing the Mechanism of Action. Target. Oncol. 2017, 12, 19–35. [Google Scholar] [CrossRef]
- Makhov, P.; Naito, S.; Haifler, M.; Kutikov, A.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. The convergent roles of NF-κB and ER stress in sunitinib-mediated expression of pro-tumorigenic cytokines and refractory phenotype in renal cell carcinoma. Cell Death Dis. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, G.; Che, X.; Li, Q.; Zhang, Z.; Tang, Q. Sorafenib induces renal cell carcinoma apoptosis via upregulating activating transcription factor 4. Pharm. Int. J. Pharm. Sci. 2018, 73, 156–160. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Sierra, T.; Bellido, B.; Reyes-Fermín, L.M.; Martínez-Klimova, E.; Pedraza-Chaverri, J. Regulation of endoplasmic reticulum stress in models of kidney disease. Adv. Redox Res. 2021, 3, 100010. [Google Scholar] [CrossRef]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Sheshadri, N.; Poria, D.K.; Sharan, S.; Hu, Y.; Yan, C.; Koparde, V.N.; Balamurugan, K.; Sterneck, E. PERK signaling through C/EBPδ contributes to ER stress-induced expression of immunomodulatory and tumor promoting chemokines by cancer cells. Cell Death Dis. 2021, 12, 1038. [Google Scholar] [CrossRef]
- Fu, X.; Cui, J.; Meng, X.; Jiang, P.; Zheng, Q.; Zhao, W.; Chen, X. Endoplasmic reticulum stress, cell death and tumor: Association between endoplasmic reticulum stress and the apoptosis pathway in tumors. Oncol. Rep. 2021, 45, 801–808. [Google Scholar] [CrossRef]
- Pagliarini, V.; Giglio, P.; Bernardoni, P.; De Zio, D.; Fimia, G.M.; Piacentini, M.; Corazzari, M. Downregulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 2015, 128, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Peired, A.J.; Lazzeri, E.; Guzzi, F.; Anders, H.-J.; Romagnani, P. From kidney injury to kidney cancer. Kidney Int. 2021, 100, 55–66. [Google Scholar] [CrossRef]
- Renal Cell Cancer and Chronic Kidney Disease|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1548559521001294?token=5622926FC9B96AC5789EAC0796CE4D83E1AC96B61442CD43B6C0244C3BFFB5A485CE17C93085F09DA0823EA59E091EA0&originRegion=eu-west-1&originCreation=20221202090532 (accessed on 2 December 2022).
- Yamaguchi, J.; Tanaka, T.; Nangaku, M. Recent advances in understanding of chronic kidney disease. F1000Research 2015, 4, 1212. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Sanchez, D.J.; Simon, M.C. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The Evolving Understanding of the Contribution of Lipid Metabolism to Diabetic Kidney Disease. Curr. Diab. Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Lee, K.; Wang, N.; He, J.C. The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy. Curr. Diab. Rep. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Yuan, C.; Wu, X. Endoplasmic Reticulum Stress in Diabetic Nephrology: Regulation, Pathological Role, and Therapeutic Potential. Oxid. Med. Cell. Longev. 2021, 2021, 7277966. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Zhu, N.; Luo, F.; Xu, Y.; Fang, X. Targeted inhibition of endoplasmic reticulum stress: New hope for renal fibrosis (Review). Mol. Med. Rep. 2017, 16, 1014–1020. [Google Scholar] [CrossRef] [Green Version]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef]
- Gallazzini, M.; Pallet, N. Endoplasmic reticulum stress and kidney dysfunction. Biol. Cell 2018, 110, 205–216. [Google Scholar] [CrossRef]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef]
- Elnagar, G.M.; Elseweidy, M.M.; Elkomy, N.M.I.M.; Al-Gabri, N.A.; Shawky, M. 10-Dehydrogingerdione ameliorates renal endoplasmic reticulum/oxidative stress and apoptosis in alcoholic nephropathy induced in experimental rats. Life Sci. 2021, 279, 119673. [Google Scholar] [CrossRef]
- Kemter, E.; Fröhlich, T.; Arnold, G.J.; Wolf, E.; Wanke, R. Mitochondrial Dysregulation Secondary to Endoplasmic Reticulum Stress in Autosomal Dominant Tubulointerstitial Kidney Disease—UMOD (ADTKD-UMOD). Sci. Rep. 2017, 7, 42970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jilg, C.A.; Drendel, V.; Bacher, J.; Pisarski, P.; Neeff, H.; Drognitz, O.; Schwardt, M.; Gläsker, S.; Malinoc, A.; Erlic, Z.; et al. Autosomal dominant polycystic kidney disease: Prevalence of renal neoplasias in surgical kidney specimens. Nephron Clin. Pr. 2013, 123, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhang, W.; Wen, Y.; Sun, Y.; Chen, L.; Li, H.; Li, M.; Li, X.; Lafayette, R.A.; Li, X. Endoplasmic Reticulum Stress Predicts Clinical Response to Cyclosporine Treatment in Primary Membranous Nephropathy. Am. J. Nephrol. 2016, 43, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V.; Takano, T.; Papillon, J.; Bijian, K. Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury. J. Biol. Chem. 2005, 280, 24396–24403. [Google Scholar] [CrossRef] [Green Version]
- Jehn, U.; Bayraktar, S.; Pollmann, S.; Van Marck, V.; Weide, T.; Pavenstädt, H.; Brand, E.; Lenders, M. α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. Int. J. Mol. Sci. 2021, 22, 11339. [Google Scholar] [CrossRef]
- Boesen, E.I.; Bonnemaison, M.L. Endoplasmic Reticulum Stress Promotes the Development and Progression of Lupus Nephritis. FASEB J. 2018, 32, 849.16. [Google Scholar] [CrossRef]
- Tseng, C.-H. Type 2 Diabetes Mellitus and Kidney Cancer Risk: A Retrospective Cohort Analysis of the National Health Insurance. PLoS ONE 2015, 10, e0142480. [Google Scholar] [CrossRef]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Renal Physiol. 2010, 299, F821–F829. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Hao, Y.; Li, H.; Liu, Q.; Gao, F.; Liu, W.; Duan, H. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int. J. Mol. Med. 2014, 33, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, J.; Pan, T.; Chen, X.; Xu, X.; Jiang, D.; Yin, J. Role of the ER stress in prostaglandin E2/E-prostanoid 2 receptor involved TGF-β1-induced mice mesangial cell injury. Mol. Cell. Biochem. 2016, 411, 43–55. [Google Scholar] [CrossRef]
- Park, M.-J.; Han, H.J.; Kim, D.-I. Lipotoxicity-Induced PRMT1 Exacerbates Mesangial Cell Apoptosis via Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2017, 18, E1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.; Kuo, M.-C.; Hung, W.-W.; Wu, L.-Y.; Wu, P.-H.; Chang, W.-A.; Kuo, P.-L.; Hsu, Y.-L. High Glucose Induces Mesangial Cell Apoptosis through miR-15b-5p and Promotes Diabetic Nephropathy by Extracellular Vesicle Delivery. Mol. Ther. 2020, 28, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dong, X.-J.; Ding, M.-R.; You, C.-Y.; Lin, X.; Wang, Y.; Wu, M.-J.; Xu, G.-F.; Wang, G.-D. Resveratrol decreases high glucose-induced apoptosis in renal tubular cells via suppressing endoplasmic reticulum stress. Mol. Med. Rep. 2020, 22, 4367–4375. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.-R.; Chen, X.-M.; Cai, G.-Y.; Lin, L.-R.; He, Y.-N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol. Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef] [Green Version]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in renal proximal tubular cells: Relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef]
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res. Int. 2021, 2021, 1497449. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.S.; Ponnusamy, L.; Singh, K.P. Chronic Oxidative Stress Leads to Malignant Transformation Along With Acquisition of Stem Cell Characteristics, and Epithelial to Mesenchymal Transition in Human Renal Epithelial Cells. J. Cell. Physiol. 2015, 230, 1916–1928. [Google Scholar] [CrossRef]
- Labochka, D.; Moszczuk, B.; Kukwa, W.; Szczylik, C.; Czarnecka, A.M. Mechanisms through which diabetes mellitus influences renal cell carcinoma development and treatment: A review of the literature. Int. J. Mol. Med. 2016, 38, 1887–1894. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Yu, J.; Yu, F.; Yang, S.; Qian, Q.; Zha, Y. IGF-1/IGF-1R blockade ameliorates diabetic kidney disease through normalizing Snail1 expression in a mouse model. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E686–E698. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Huang, H.; Tan, L.; Wan, J.; Gui, H.; Zhao, L.; Ruan, X.; Chen, X.; Du, X. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobulescu, I.A.; Pop, L.M.; Mani, C.; Turner, K.; Rivera, C.; Khatoon, S.; Kairamkonda, S.; Hannan, R.; Palle, K. Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma. Metabolites 2021, 11, 608. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Monteillet, L.; Calderaro, J.; Mithieux, G.; Rajas, F. Polycystic kidney features of the renal pathology in glycogen storage disease type I: Possible evolution to renal neoplasia. J. Inherit. Metab. Dis. 2018, 41, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Monteillet, L.; Gjorgjieva, M.; Silva, M.; Verzieux, V.; Imikirene, L.; Duchampt, A.; Guillou, H.; Mithieux, G.; Rajas, F. Intracellular Lipids are an Independent Cause of Liver Injury and Chronic Kidney Disease in Non Alcoholic Fatty Liver Disease-like Context. Mol. Metab. 2018, 16, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Raffin, M.; Duchampt, A.; Perry, A.; Stefanutti, A.; Brevet, M.; Tortereau, A.; Dubourg, L.; Hubert-Buron, A.; Mabille, M.; et al. Progressive development of renal cysts in glycogen storage disease type I. Hum. Mol. Genet. 2016, 25, 3784–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.-X.; Zhu, H.-H.; Zhu, Y.-M. Diabetes and cancer: Associations, mechanisms, and implications for medical practice. World J. Diabetes 2014, 5, 372–380. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, K.; Ji, W.; Zhang, W.; Li, Y.; Liu, M.; Cui, J.; Li, W. Effects of Diabetes on Inflammatory Status and Prognosis in Cancer Patients. Front. Nutr. 2022, 9, 792577. [Google Scholar] [CrossRef]
- Foresto-Neto, O.; Albino, A.H.; Arias, S.C.A.; Faustino, V.D.; Zambom, F.F.F.; Cenedeze, M.A.; Elias, R.M.; Malheiros, D.M.A.C.; Camara, N.O.S.; Fujihara, C.K.; et al. NF-κB System Is Chronically Activated and Promotes Glomerular Injury in Experimental Type 1 Diabetic Kidney Disease. Front. Physiol. 2020, 11, 84. [Google Scholar] [CrossRef]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J. Cell. Physiol. 2019, 234, 3043–3057. [Google Scholar] [CrossRef]
- Hofherr, A.; Williams, J.; Gan, L.-M.; Söderberg, M.; Hansen, P.B.L.; Woollard, K.J. Targeting inflammation for the treatment of Diabetic Kidney Disease: A five-compartment mechanistic model. BMC Nephrol. 2022, 23, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, Q.; Liu, B.; Ma, Q.; Zhang, T.; Huang, T.; Lv, Z.; Wang, R. Chronic Kidney Disease and Cancer: Inter-Relationships and Mechanisms. Front. Cell Dev. Biol. 2022, 10, 868715. [Google Scholar] [CrossRef] [PubMed]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. Unexpected relevance of the hallmarks of cancer to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çeker, T.; Yılmaz, Ç.; Kırımlıoglu, E.; Aslan, M. Endoplasmic-reticulum-stress-induced lipotoxicity in human kidney epithelial cells. Toxicol. Res. 2022, 11, 683–695. [Google Scholar] [CrossRef]
- Kuo, C.-Y.; Lin, C.-H.; Hsu, T. VHL Inactivation in Precancerous Kidney Cells Induces an Inflammatory Response via ER Stress-Activated IRE1α Signaling. Cancer Res. 2017, 77, 3406–3416. [Google Scholar] [CrossRef] [Green Version]
- Salvagno, C.; Mandula, J.K.; Rodriguez, P.C.; Cubillos-Ruiz, J.R. Decoding endoplasmic reticulum stress signals in cancer cells and antitumor immunity. Trends Cancer 2022, 8, 930–943. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Liang, Y.; Qu, L.; Liu, Z.; Liang, L.; Wang, Y.; Quan, S.; Wang, Y.; Tang, L. The IRE1/JNK signaling pathway regulates inflammation cytokines and production of glomerular extracellular matrix in the acute kidney injury to chronic kidney disease transition. Mol. Biol. Rep. 2022, 49, 7709–7718. [Google Scholar] [CrossRef]
- Bao, Y.; Liang, W.; Ye, Y.; Yi, B. PERK-Dependent Activation of the JAK2/STAT3 Pathway Contributes to High Glucose-Induced Extracellular Matrix Deposition in Renal Tubular Epithelial Cells. Int. J. Endocrinol. 2021, 2021, 8475868. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Shu, S.; Wang, H.; Zhu, J.; Liu, Z.; Yang, D.; Wu, W.; Cai, J.; Chen, A.; Tang, C.; Dong, Z. Reciprocal regulation between ER stress and autophagy in renal tubular fibrosis and apoptosis. Cell Death Dis. 2021, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tian, G. Autophagy as a Vital Therapy Target for Renal Cell Carcinoma. Front. Pharmacol. 2021, 11, 518225. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Cruz, M.S.; Simmen, T. Cancer: Untethering Mitochondria from the Endoplasmic Reticulum? Front. Oncol. 2017, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Yang, W.; Sun, L. Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Prospective Roles in Kidney Disease. Oxid. Med. Cell. Longev. 2020, 2020, 3120539. [Google Scholar] [CrossRef]
- Wei, X.; Wei, X.; Lu, Z.; Li, L.; Hu, Y.; Sun, F.; Jiang, Y.; Ma, H.; Zheng, H.; Yang, G.; et al. Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism. 2020, 105, 154182. [Google Scholar] [CrossRef]
- Yang, M.; Zhao, L.; Gao, P.; Zhu, X.; Han, Y.; Chen, X.; Li, L.; Xiao, Y.; Wei, L.; Li, C.; et al. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAM integrity. EBioMedicine 2019, 43, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, X.; Wei, X.; Feng, H.; Hu, B.; Deng, Z.; Liu, B.; Luan, Y.; Ruan, Y.; Liu, X.; et al. A Mitochondrial Dysfunction and Oxidative Stress Pathway-Based Prognostic Signature for Clear Cell Renal Cell Carcinoma. Oxid. Med. Cell. Longev. 2021, 2021, 9939331. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, H.; Lindgren, D.; Mandahl Forsberg, A.; Mulder, H.; Axelson, H.; Johansson, M.E. Primary clear cell renal carcinoma cells display minimal mitochondrial respiratory capacity resulting in pronounced sensitivity to glycolytic inhibition by 3-Bromopyruvate. Cell Death Dis. 2015, 6, e1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathmell, W.K.; Rathmell, J.C.; Linehan, W.M. Metabolic Pathways in Kidney Cancer: Current Therapies and Future Directions. J. Clin. Oncol. 2018, 36, 3540–3546. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldewijns, M.M.; van Vlodrop, I.J.H.; Vermeulen, P.B.; Soetekouw, P.M.M.B.; van Engeland, M.; de Bruïne, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.A.; Paffrath, V.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation. Cancers 2019, 11, 1298. [Google Scholar] [CrossRef] [Green Version]
- Krencz, I.; Vetlényi, E.; Dankó, T.; Petővári, G.; Moldvai, D.; Sztankovics, D.; Raffay, R.; Mészáros, K.; Sebestyén, E.; Végső, G.; et al. Metabolic Adaptation as Potential Target in Papillary Renal Cell Carcinomas Based on Their In Situ Metabolic Characteristics. Int. J. Mol. Sci. 2022, 23, 10587. [Google Scholar] [CrossRef]
- Shin Lee, J.; Seok Kim, H.; Bok Kim, Y.; Cheol Lee, M.; Soo Park, C. Expression of PTEN in renal cell carcinoma and its relation to tumor behavior and growth. J. Surg. Oncol. 2003, 84, 166–172. [Google Scholar] [CrossRef]
- Lu, H.; Tan, Y.; Chen, L. A clinical study on the expression of PTEN in renal cell carcinoma in children. Oncol. Lett. 2019, 17, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumor-Associated Mutant p53 Drives the Warburg Effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- van der Harg, J.M.; van Heest, J.C.; Bangel, F.N.; Patiwael, S.; van Weering, J.R.T.; Scheper, W. The UPR reduces glucose metabolism via IRE1 signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Jao, T.-M.; Nangaku, M.; Wu, C.-H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.-Q. The UPR Transducer IRE1 Promotes Breast Cancer Malignancy by Degrading Tumor Suppressor microRNAs. Iscience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic Reticulum Stress and Cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, M.; Koyanagi, S.; Okamoto, A.; Suzuki, S.O.; Matsunaga, N.; Ohdo, S. Stress-regulated transcription factor ATF4 promotes neoplastic transformation by suppressing expression of the INK4a/ARF cell senescence factors. Cancer Res. 2012, 72, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Romeo, M.A.; Arena, A.; Gilardini Montani, M.S.; Di Renzo, L.; D’Orazi, G.; Cirone, M. ATF6 prevents DNA damage and cell death in colon cancer cells undergoing ER stress. Cell Death Discov. 2022, 8, 295. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Hsu, C.-C.; Huang, T.-T.; Lee, C.-H.; Chen, J.-L.; Yang, S.-H.; Jiang, J.-K.; Chen, W.-S.; Lee, K.-D.; Teng, H.-W. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 2018, 12, 1706–1717. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.; West, C.; Moore, R.P.; Hoff, D.V.; Dent, P. GZ17-6.02 and axitinib interact to kill renal carcinoma cells. Oncotarget 2022, 13, 281–290. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Marlow, L.A.; Kennedy, W.P.; Kennedy, G.T.; Copland, J.A.; Menefee, M.E. Preclinical evaluation of the mTOR inhibitor, temsirolimus, in combination with the epothilone B analog, ixabepilone in renal cell carcinoma. Am. J. Cancer Res. 2013, 3, 390–401. [Google Scholar]
- Okubo, K.; Isono, M.; Asano, T.; Sato, A. Panobinostat and Nelfinavir Inhibit Renal Cancer Growth by Inducing Endoplasmic Reticulum Stress. Anticancer Res. 2018, 38, 5615–5626. [Google Scholar] [CrossRef]
- Okubo, K.; Isono, M.; Miyai, K.; Asano, T.; Sato, A. Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells. Cancer Sci. 2020, 111, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Sato, A.; Asano, T.; Okubo, K.; Asano, T. Delanzomib Interacts with Ritonavir Synergistically to Cause Endoplasmic Reticulum Stress in Renal Cancer Cells. Anticancer Res. 2018, 38, 3493–3500. [Google Scholar] [CrossRef]
- Isono, M.; Sato, A.; Okubo, K.; Asano, T.; Asano, T. Ritonavir Interacts With Belinostat to Cause Endoplasmic Reticulum Stress and Histone Acetylation in Renal Cancer Cells. Oncol. Res. 2016, 24, 327–335. [Google Scholar] [CrossRef]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef]
- Batova, A.; Altomare, D.; Creek, K.E.; Naviaux, R.K.; Wang, L.; Li, K.; Green, E.; Williams, R.; Naviaux, J.C.; Diccianni, M.; et al. Englerin A induces an acute inflammatory response and reveals lipid metabolism and ER stress as targetable vulnerabilities in renal cell carcinoma. PLoS ONE 2017, 12, e0172632. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.J.; Park, E.J.; Min, K.J.; Park, J.-W.; Kwon, T.K. Endoplasmic reticulum stress mediates withaferin A-induced apoptosis in human renal carcinoma cells. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2011, 25, 692–698. [Google Scholar] [CrossRef]
- Zhai, X.; Yuan, S.; Yang, X.; Zou, P.; Li, L.; Li, G.; Shao, Y.; Abd El-Aty, A.M.; Hacımüftüoğlu, A.; Wang, J. Chitosan Oligosaccharides Induce Apoptosis in Human Renal Carcinoma via Reactive-Oxygen-Species-Dependent Endoplasmic Reticulum Stress. J. Agric. Food Chem. 2019, 67, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zhuo, R.; Dai, J.; Wang, X.; Huang, X.; Wang, H.; Xu, D. Chelerythrine induces apoptosis via ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human renal cell carcinoma. J. Cell. Mol. Med. 2020, 24, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zheng, W.; Rong, L.; Xing, Y.; Hu, D. Bicyclol exerts an anti-tumor effect via ROS-mediated endoplasmic reticulum stress in human renal cell carcinoma cells. Biomed. Pharmacother. 2017, 91, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Jung, K.-J.; Kwon, T.K. Carnosic Acid Induces Apoptosis Through Reactive Oxygen Species-mediated Endoplasmic Reticulum Stress Induction in Human Renal Carcinoma Caki Cells. J. Cancer Prev. 2014, 19, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-H.; Chiou, H.-L.; Lin, C.-L.; Lin, C.-Y.; Yang, S.-F.; Hsieh, Y.-H. Induction of endoplasmic reticulum stress and mitochondrial dysfunction dependent apoptosis signaling pathway in human renal cancer cells by norcantharidin. Oncotarget 2018, 9, 4787–4797. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Min, K.; Kim, S.; Park, J.-W.; Kwon, T.K. RU486 Induces Pro-Apoptotic Endoplasmic Reticulum Stress Through the Induction of CHOP Expression by Enhancing C/EBPδ Expression in Human Renal Carcinoma Caki Cells. J. Cell. Biochem. 2016, 117, 361–369. [Google Scholar] [CrossRef]
- Lee, C.-H.; Hung, P.-F.; Lu, S.-C.; Chung, H.-L.; Chiang, S.-L.; Wu, C.-T.; Chou, W.-C.; Sun, C.-Y. MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis. Int. J. Mol. Sci. 2019, 20, 6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Allemailem, K.S.; Almatroudi, A.; Almatroodi, S.A.; Mahzari, A.; Alsahli, M.A.; Rahmani, A.H. Endoplasmic Reticulum Stress Provocation by Different Nanoparticles: An Innovative Approach to Manage the Cancer and Other Common Diseases. Molecules 2020, 25, 5336. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Hou, M.; Zhang, S.; Xin, Z.; Huang, J.; Yang, J.; Wang, Y.; Cai, X.; Xie, S.; Zhang, C.; et al. Dual Targeting of Endoplasmic Reticulum by Redox-Deubiquitination Regulation for Cancer Therapy. Int. J. Nanomed. 2021, 16, 5193–5209. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, Y.; Yang, Q.; Gao, Y.; Duan, X.; Fu, Q.; Chu, C.; Pan, X.; Cui, X.; Sun, Y. Cuprous oxide nanoparticles trigger ER stress-induced apoptosis by regulating copper trafficking and overcoming resistance to sunitinib therapy in renal cancer. Biomaterials 2017, 146, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Han, K.S.; Li, N.; Raven, P.A.; Fazli, L.; Frees, S.; Ettinger, S.; Park, K.C.; Hong, S.J.; Gleave, M.E.; So, A.I. Inhibition of endoplasmic reticulum chaperone protein glucose-regulated protein 78 potentiates anti-angiogenic therapy in renal cell carcinoma through inactivation of the PERK/eIF2α pathway. Oncotarget 2015, 6, 34818–34830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Gao, Y.; Liu, A.; Yang, X.; Huang, F.; Xu, L.; Danfeng, X.; Chen, L. EIF3D promotes sunitinib resistance of renal cell carcinoma by interacting with GRP78 and inhibiting its degradation. EbioMedicine 2019, 49, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Li, Z.; Han, W.; Zhu, C.; Lou, N.; Li, X.; Luo, G.; Peng, S.; Li, G.; Zhao, Y.; et al. Low DAPK1 expression correlates with poor prognosis and sunitinib resistance in clear cell renal cell carcinoma. Aging 2020, 13, 1842–1858. [Google Scholar] [CrossRef]
- Han, K.S.; Li, N.; Raven, P.A.; Li, E.; Fazli, L.; Gleave, M.E.; So, A.I. ER stress protein GRP78 as a therapeutic target combined with antiangiogenic therapy in renal cell carcinoma. J. Clin. Oncol. 2013, 31, 428. [Google Scholar] [CrossRef]
- Matsumura, K.; Sakai, C.; Kawakami, S.; Yamashita, F.; Hashida, M. Inhibition of cancer cell growth by GRP78 siRNA lipoplex via activation of unfolded protein response. Biol. Pharm. Bull. 2014, 37, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Sobczuk, P.; Brodziak, A.; Khan, M.I.; Chhabra, S.; Fiedorowicz, M.; Wełniak-Kamińska, M.; Synoradzki, K.; Bartnik, E.; Cudnoch-Jędrzejewska, A.; Czarnecka, A.M. Choosing The Right Animal Model for Renal Cancer Research. Transl. Oncol. 2020, 13, 100745. [Google Scholar] [CrossRef]
- Nyga, A.; Stamati, K.; Redondo, P.A.; Azimi, T.; Feber, A.; Neves, J.B.; Hamoudi, R.; Presneau, N.; El Sheikh, S.; Tran, M.G.B.; et al. Renal tumouroids: Challenges of manufacturing 3D cultures from patient derived primary cells. J. Cell Commun. Signal. 2022, 16, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Batchelder, C.A.; Martinez, M.L.; Duru, N.; Meyers, F.J.; Tarantal, A.F. Three Dimensional Culture of Human Renal Cell Carcinoma Organoids. PLoS ONE 2015, 10, e0136758. [Google Scholar] [CrossRef] [PubMed]
- Esser, L.K.; Branchi, V.; Leonardelli, S.; Pelusi, N.; Simon, A.G.; Klümper, N.; Ellinger, J.; Hauser, S.; Gonzalez-Carmona, M.A.; Ritter, M.; et al. Cultivation of Clear Cell Renal Cell Carcinoma Patient-Derived Organoids in an Air-Liquid Interface System as a Tool for Studying Individualized Therapy. Front. Oncol. 2020, 10, 01775. [Google Scholar] [CrossRef] [PubMed]
- Kazama, A.; Anraku, T.; Kuroki, H.; Shirono, Y.; Murata, M.; Bilim, V.; Ugolkov, A.; Saito, K.; Tomita, Y. Development of patient-derived tumor organoids and a drug testing model for renal cell carcinoma. Oncol. Rep. 2021, 46, 8177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Garg, H.; Gupta, N.; Sharma, A.; Kaushal, S.; Kumar, R.; Dinda, A.K. Glucose- regulated protein 78 (GRP78) in renal cell carcinoma: A novel biomarker for predicting tumor behavior. Heliyon 2021, 7, e07300. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Cohen, M. Linking cell-surface GRP78 to cancer: From basic research to clinical value of GRP78 antibodies. Cancer Lett. 2022, 524, 1–14. [Google Scholar] [CrossRef]
- Ni, M.; Zhou, H.; Wey, S.; Baumeister, P.; Lee, A.S. Regulation of PERK Signaling and Leukemic Cell Survival by a Novel Cytosolic Isoform of the UPR Regulator GRP78/BiP. PLoS ONE 2009, 4, e6868. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Song, Y.; Dai, J.; Wang, Z.; Zeng, Y.; Chen, F.; Zhang, P. Endoplasmic Reticulum Stress-Related Signature Predicts Prognosis and Drug Response in Clear Cell Renal Cell Carcinoma. Front. Pharmacol. 2022, 13, 909123. [Google Scholar] [CrossRef]
- Shen, Y.; Cao, Y.; Zhou, L.; Wu, J.; Mao, M. Construction of an endoplasmic reticulum stress-related gene model for predicting prognosis and immune features in kidney renal clear cell carcinoma. Front. Mol. Biosci. 2022, 9, 928006. [Google Scholar] [CrossRef]
- Du, Z.; Chen, W.; Xia, Q.; Shi, O.; Chen, Q. Trends and projections of kidney cancer incidence at the global and national levels, 1990–2030: A Bayesian age-period-cohort modeling study. Biomark. Res. 2020, 8, 16. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia de Sousa, M.; Delangre, E.; Türkal, M.; Foti, M.; Gjorgjieva, M. Endoplasmic Reticulum Stress in Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 4914. https://doi.org/10.3390/ijms24054914
Correia de Sousa M, Delangre E, Türkal M, Foti M, Gjorgjieva M. Endoplasmic Reticulum Stress in Renal Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(5):4914. https://doi.org/10.3390/ijms24054914
Chicago/Turabian StyleCorreia de Sousa, Marta, Etienne Delangre, Miranda Türkal, Michelangelo Foti, and Monika Gjorgjieva. 2023. "Endoplasmic Reticulum Stress in Renal Cell Carcinoma" International Journal of Molecular Sciences 24, no. 5: 4914. https://doi.org/10.3390/ijms24054914