
The Role of NLRP3, a Star of Excellence in Myeloproliferative Neoplasms

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
Abstract
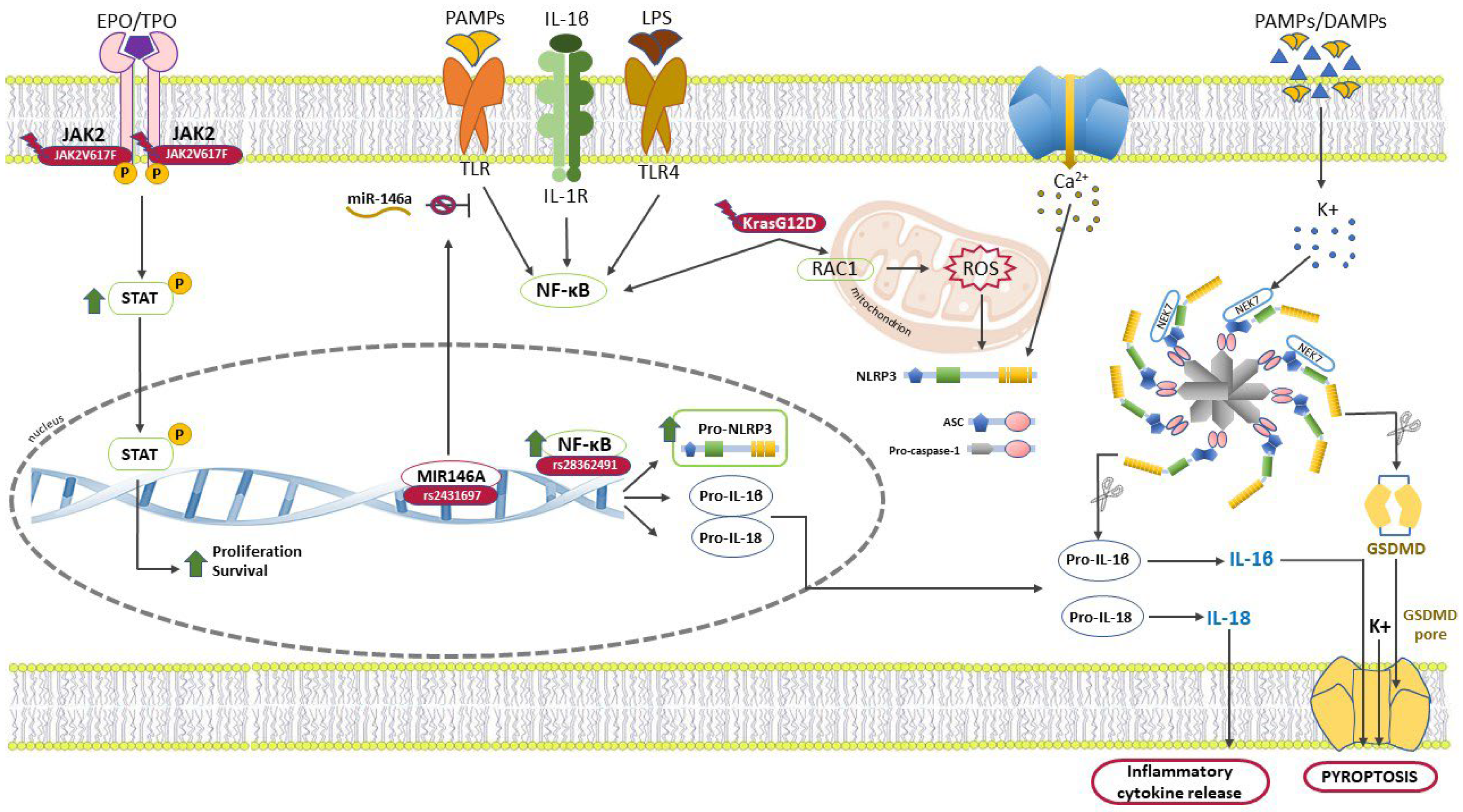
:1. Introduction
2. Myeloproliferative Neoplasms: The Inflammatory and Immune Environment
3. NLRP3 Protein

4. NLRP3, the Keystone in MPNs
5. Novel Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corey, S.J.; Minden, M.D.; Barber, D.L.; Kantarjian, H.; Wang, J.C.Y.; Schimmer, A.D. Myelodysplastic Syndromes: The Complexity of Stem-Cell Diseases. Nat. Rev. Cancer 2007, 7, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L.; Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 12, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic Inflammation as a Promotor of Mutagenesis in Essential Thrombocythemia, Polycythemia Vera and Myelofibrosis. A Human Inflammation Model for Cancer Development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of Bone Marrow Hematopoietic Stem Cell Niche. Int. J. Hematol. 2017, 106, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic Basis and Molecular Pathophysiology of Classical Myeloproliferative Neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Casieri, P.; Rossi, A.R.; Vicari, L.; Liso, V.; Rocchi, M.; Specchia, G. Non Random Distribution of Genomic Features in Breakpoint Regions Involved in Chronic Myeloid Leukemia Cases with Variant t(9;22) or Additional Chromosomal Rearrangements. Mol. Cancer 2010, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; D’Addabbo, P.; Liso, V.; Rocchi, M.; Specchia, G. Genomic Segmental Duplications on the Basis of the t(9;22) Rearrangement in Chronic Myeloid Leukemia. Oncogene 2010, 29, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Apperley, J.F. Chronic Myeloid Leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired Mutation of the Tyrosine Kinase JAK2 in Human Myeloproliferative Disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Nangalia, J.; Green, A.R.; Vannucchi, A.M. CALR Mutations in Myeloproliferative Neoplasms: Hidden behind the Reticulum. Am. J. Hematol. 2014, 89, 453–456. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-Mutated or Triple-Negative Myelofibrosis: Clinical, Cytogenetic and Molecular Comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Green, A.R. Molecular Determinants of Pathogenesis and Clinical Phenotype in Myeloproliferative Neoplasms. Haematologica 2017, 102, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted Deep Sequencing in Primary Myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal Evolution and Clinical Correlates of Somatic Mutations in Myeloproliferative Neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Andina, N.; Bonadies, N.; Allam, R. Inflammasome Activation in Myeloid Malignancies-Friend or Foe? Front. Cell Dev. Biol. 2022, 9, 825611. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 Inflammasome as a New Therapeutic Option for Overcoming Cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, M.E.; Suman, R.; Harrison, H.J.; Jackson, G.E.; Howard, M.R.; Hitchcock, I.S. Endothelial JAK2V617F Expression Drives Inflammation and Cellular Senescence; New Evidence for the Roles of Endothelial Cells in MPN-Related Clotting Abnormalities? Blood 2016, 128, 3134. [Google Scholar] [CrossRef]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of Inflammation on Myeloproliferative Neoplasm Symptom Development. Mediat. Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- de Mooij, C.E.M.; Netea, M.G.; van der Velden, W.J.F.M.; Blijlevens, N.M.A. Targeting the Interleukin-1 Pathway in Patients with Hematological Disorders. Blood 2017, 129, 3155–3164. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of Haematopoietic Stem Cell Niche Is Essential for Myeloproliferative Neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Rai, S.; Hansen, N.; Hao-Shen, H.; Dirnhofer, S.; Tata, N.R.; Skoda, R.C. IL-1β Secreted from Mutant Cells Carrying JAK2-V617Ffavors Early Clonal Expansion and Promotes MPN Disease Initiation and Progression. Blood 2019, 134, 307. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative Neoplasms and Inflammation: Whether to Target the Malignant Clone or the Inflammatory Process or Both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT Pathway Activation in Malignant and Nonmalignant Cells Contributes to MPN Pathogenesis and Therapeutic Response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective Negative Regulation of Toll-like Receptor Signaling Leads to Excessive TNF-α in Myeloproliferative Neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zuo, X. Cytokines Frequently Implicated in Myeloproliferative Neoplasms. Cytokine X 2019, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Rambaldi, A. Inflammation and Myeloproliferative Neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 Inflammasome and Its Inhibitors: A Review. Front. Pharm. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic Immune Response Dysregulation in MDS Pathogenesis. Blood 2018, 132, 1553. [Google Scholar] [CrossRef] [Green Version]
- Gañán-Gómez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of Innate Immune and Inflammatory Signaling in Myelodysplastic Syndromes. Leukemia 2015, 29, 1458. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Kanneganti, T.D. Diverging Inflammasome Signals in Tumorigenesis and Potential Targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Heilig, R.; Broz, P. Function and Mechanism of the Pyrin Inflammasome. Eur. J. Immunol. 2018, 48, 230–238. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kanneganti, T.D. Humans PIKK-up NLRP3 to Skip NEK7. Trends Immunol. 2022, 43, 947–949. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1β–Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in Health and Disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Shao, Y.; Cheng, Z.; Li, X.; Chernaya, V.; Wang, H.; Yang, X.F. Immunosuppressive/Anti-Inflammatory Cytokines Directly and Indirectly Inhibit Endothelial Dysfunction-A Novel Mechanism for Maintaining Vascular Function. J. Hematol. Oncol. 2014, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C. The Role of Inflammatory Cytokines in Endothelial Dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.K.; Kucia, M. The Nlrp3 Inflammasome as a “Rising Star” in Studies of Normal and Malignant Hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Kawashima, Y.; Kurima, K.; Chae, J.J.; Ross, A.M.; Pinto-Patarroyo, G.; Patel, S.K.; Muskett, J.A.; Ratay, J.S.; Chattaraj, P.; et al. NLRP3 Mutation and Cochlear Autoinflammation Cause Syndromic and Nonsyndromic Hearing Loss DFNA34 Responsive to Anakinra Therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E7766–E7775. [Google Scholar] [CrossRef]
- Yang, G.; Lee, H.E.; Moon, S.J.; Ko, K.M.; Koh, J.H.; Seok, J.K.; Min, J.K.; Heo, T.H.; Kang, H.C.; Cho, Y.Y.; et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol. 2020, 72, 1192–1202. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.D. Recent Advances In Inflammasome Biology. Curr. Opin. Immunol. 2018, 50, 32. [Google Scholar] [CrossRef] [PubMed]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 Inflammasome Pathway. J. Inflamm. Res. 2018, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A New Frontier in Cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. NLRP3: A Novel Mediator in Cardiovascular Disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; al Ali, N.H.; Padron, E.; et al. Oxidized Mitochondrial DNA Released after Inflammasome Activation Is a Disease Biomarker for Myelodysplastic Syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.B.; Opitz, B.; van der Meer, J.H.M.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential Requirement for the Activation of the Inflammasome for Processing and Release of IL-1beta in Monocytes and Macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, G.; Dinarello, C.A. Interleukin-18 and Interleukin-1 Beta: Two Cytokine Substrates for ICE (Caspase-1). J. Clin. Immunol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 Mediates Non-Canonical Activation of the NLRP3 Inflammasome in Human Myeloid Cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef]
- Chu, L.H.; Indramohan, M.; Ratsimandresy, R.A.; Gangopadhyay, A.; Morris, E.P.; Monack, D.M.; Dorfleutner, A.; Stehlik, C. The Oxidized Phospholipid OxPAPC Protects from Septic Shock by Targeting the Non-Canonical Inflammasome in Macrophages. Nat. Commun. 2018, 9, 996. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic Flagellin Activates Caspase-1 and Secretion of Interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Lenkiewicz, A.M.; Adamiak, M.; Thapa, A.; Bujko, K.; Pedziwiatr, D.; Abdel-Latif, A.K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Nlrp3 Inflammasome Orchestrates Mobilization of Bone Marrow-Residing Stem Cells into Peripheral Blood. Stem Cell Rev. 2019, 15, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Adamiak, M.; Plonka, M.; Abdel-Latif, A.; Ratajczak, J. Mobilization of Hematopoietic Stem Cells as a Result of Innate Immunity-Mediated Sterile Inflammation in the Bone Marrow Microenvironment—The Involvement of Extracellular Nucleotides and Purinergic Signaling. Leukemia 2018, 32, 1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamiak, M.; Abdelbaset-Ismail, A.; Suszynska, M.; Abdel-Latif, A.; Ratajczak, J.; Ratajczak, M.Z. Novel Evidence That the Mannan-Binding Lectin Pathway of Complement Activation Plays a Pivotal Role in Triggering Mobilization of Hematopoietic Stem/Progenitor Cells by Activation of Both the Complement and Coagulation Cascades. Leukemia 2017, 31, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, A.; Masters, S.L. Homeostasis-Altering Molecular Processes as Mechanisms of Inflammasome Activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. MTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbore, G.; Kemper, C. A Novel “Complement-Metabolism-Inflammasome Axis” as a Key Regulator of Immune Cell Effector Function. Eur. J. Immunol. 2016, 46, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 Is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmacke, N.A.; O’Duill, F.; Gaidt, M.M.; Szymanska, I.; Kamper, J.M.; Schmid-Burgk, J.L.; Mädler, S.C.; Mackens-Kiani, T.; Kozaki, T.; Chauhan, D.; et al. IKKβ Primes Inflammasome Formation by Recruiting NLRP3 to the Trans-Golgi Network. Immunity 2022, 55, 2271–2284.E7. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 Restricts Spontaneous NLRP3 Activation and Cell Death to Control Myeloid Proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.A.; Wijekoon, C.P.; Liao, K.C.; Muruve, D.A. Biochemical and Structural Aspects of the ATP-Binding Domain in Inflammasome-Forming Human NLRP Proteins. IUBMB Life 2013, 65, 851–862. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward Targeting Inflammasomes: Insights into Their Regulation and Activation. Cell Res. 2020, 30, 315. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Oroz, J.; Barrera-Vilarmau, S.; Alfonso, C.; Rivas, G.; de Alba, E. ASC Pyrin Domain Self-Associates and Binds NLRP3 Protein Using Equivalent Binding Interfaces. J. Biol. Chem. 2016, 291, 19487–19501. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, C.; Anand, P.K. Right Place, Right Time: Localisation and Assembly of the NLRP3 Inflammasome. F1000Res 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2015, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-Associated Membranes (MAMs) and Inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 Inflammasome in Cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [Green Version]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 Inflammasome in Fibroblasts Links Tissue Damage with Inflammation in Breast Cancer Progression and Metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [Green Version]
- Tomasik, J.; Basak, G.W. Inflammasomes-New Contributors to Blood Diseases. Int. J. Mol. Sci. 2022, 23, 8129. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 Inflammasome Functions as a Driver of the Myelodysplastic Syndrome Phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [Green Version]
- Brydges, S.D.; Broderick, L.; McGeough, M.D.; Pena, C.A.; Mueller, J.L.; Hoffman, H.M. Divergence of IL-1, IL-18, and Cell Death in NLRP3 Inflammasomopathies. J. Clin. Investig. 2013, 123, 4695. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Yadav, R.; Rossi, R.J.; Ruby, C.E.; Weinberg, A.D.; Aguila, H.L.; Vella, A.T. IL-18 Bridges Innate and Adaptive Immunity through IFN-γ and the CD134 Pathway. J. Immunol. 2006, 177, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Jelkmann, W. Proinflammatory Cytokines Lowering Erythropoietin Production. J. Interferon. Cytokine Res. 1998, 18, 555–559. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, S.; Liu, N.; He, N.; Zhang, A.; Meng, S.; Ji, C.; Ma, D.; Ye, J. Genetic Polymorphisms and Expression of NLRP3 Inflammasome-Related Genes Are Associated with Philadelphia Chromosome-Negative Myeloproliferative Neoplasms. Hum. Immunol. 2020, 81, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, K. Myeloproliferative Neoplasms: Current Molecular Biology and Genetics. Crit. Rev. Oncol. Hematol. 2016, 98, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Yu, J.; Yan, S.; Zhao, X.; Chen, C.; Zhou, Y.; Zhao, X.; Hua, M.; Wang, R.; Zhang, C.; et al. The Genetic Polymorphism and Expression Profiles of NLRP3 Inflammasome in Patients with Chronic Myeloid Leukemia. Hum. Immunol. 2018, 79, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Neuwirt, E.; Magnani, G.; Ćiković, T.; Wöhrle, S.; Fischer, L.; Kostina, A.; Flemming, S.; Fischenich, N.J.; Saller, B.S.; Gorka, O.; et al. Tyrosine Kinase Inhibitors Can Activate the NLRP3 Inflammasome in Myeloid Cells through Lysosomal Damage and Cell Lysis. Sci. Signal. 2023, 16, 769. [Google Scholar] [CrossRef]
- Fu, D.; Zhou, J.; Zhu, W.S.; Manley, P.W.; Wang, Y.K.; Hood, T.; Wylie, A.; Xie, X.S. Imaging the Intracellular Distribution of Tyrosine Kinase Inhibitors in Living Cells with Quantitative Hyperspectral Stimulated Raman Scattering. Nat. Chem. 2014, 6, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC Transporter A3 Facilitates Lysosomal Sequestration of Imatinib and Modulates Susceptibility of Chronic Myeloid Leukemia Cell Lines to This Drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef] [Green Version]
- Villamil Giraldo, A.M.; Appelqvist, H.; Ederth, T.; Öllinger, K. Lysosomotropic Agents: Impact on Lysosomal Membrane Permeabilization and Cell Death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.-R.; Fang, W.-T.; Cheng, Z.-P.; Shen, Y.; Wang, D.-J.; Wang, Y.-Q.; Sun, L.-N. Imatinib-Induced Hepatotoxicity via Oxidative Stress and Activation of NLRP3 Inflammasome: An in Vitro and in Vivo Study. Arch. Toxicol. 2022, 96, 1075–1087. [Google Scholar] [CrossRef]
- Fisher, D.A.C.; Malkova, O.; Engle, E.K.; Miner, C.A.; Fulbright, M.C.; Behbehani, G.K.; Collins, T.B.; Bandyopadhyay, S.; Zhou, A.; Nolan, G.P.; et al. Mass Cytometry Analysis Reveals Hyperactive NF Kappa B Signaling in Myelofibrosis and Secondary Acute Myeloid Leukemia. Leukemia 2017, 31, 1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sejas, D.P.; Zhang, X.; Qiu, Y.; Nattamai, K.J.; Rani, R.; Rathbun, K.R.; Geiger, H.; Williams, D.A.; Bagby, G.C.; et al. TNF-α Induces Leukemic Clonal Evolution Ex Vivo in Fanconi Anemia Group C Murine Stem Cells. J. Clin. Investig. 2007, 117, 3283–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guralnik, J.M.; Eisenstaedt, R.S.; Ferrucci, L.; Klein, H.G.; Woodman, R.C. Prevalence of Anemia in Persons 65 Years and Older in the United States: Evidence for a High Rate of Unexplained Anemia. Blood 2004, 104, 2263–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birgegard, G.; Samuelsson, J.; Ahlstrand, E.; Ejerblad, E.; Enevold, C.; Ghanima, W.; Hasselbalch, H.; Nielsen, C.H.; Knutsen, H.; Pedersen, O.B.; et al. Inflammatory Functional Iron Deficiency Common in Myelofibrosis, Contributes to Anaemia and Impairs Quality of Life. From the Nordic MPN Study Group. Eur. J. Haematol. 2019, 102, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. MiR-146a Is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-KappaB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Aref, S.; Atia, D.; al Tamtawy, A.; al Boghdady, M.; Gouda, E. Predictive Value of MiR-146a Rs2431697 Polymorphism to Myelofibrosis Progression in Patients with Myeloproliferative Neoplasm. Asian Pac. J. Cancer Prev. 2021, 22, 3585–3589. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; de Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lübbert, M.; et al. Oncogenic KrasG12D Causes Myeloproliferation via NLRP3 Inflammasome Activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, S.; Mascarenhas, J. Novel Therapeutics in Myeloproliferative Neoplasms. J. Hematol. Oncol. 2020, 13, 162. [Google Scholar] [CrossRef]
- Mascarenhas, J. Pacritinib for the Treatment of Patients with Myelofibrosis and Thrombocytopenia. Expert. Rev. Hematol. 2022, 15, 671–684. [Google Scholar] [CrossRef]
- Bose, P.; Masarova, L.; Verstovsek, S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers 2020, 12, 2891. [Google Scholar] [CrossRef] [PubMed]
- Gigoux, M.; Holmström, M.O.; Zappasodi, R.; Park, J.J.; Pourpe, S.; Bozkus, C.C.; Mangarin, L.M.B.; Redmond, D.; Verma, S.; Schad, S.; et al. Calreticulin Mutant Myeloproliferative Neoplasms Induce MHC-I Skewing, Which Can Be Overcome by an Optimized Peptide Cancer Vaccine. Sci. Transl. Med. 2022, 14, eaba4380. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.; Buonpane, R.; Celik, H.; Marty, C.; Lei, A.; Jobe, F.; Rupar, M.; Zhang, Y.; DiMatteo, D.; Awdew, R.; et al. Discovery of INCA033989, a Monoclonal Antibody That Selectively Antagonizes Mutant Calreticulin Oncogenic Function in Myeloproliferative Neoplasms (MPNs). Blood 2022, 140, 14–15. [Google Scholar] [CrossRef]
- Yu, X.; Lv, J.; Wu, J.; Chen, Y.; Chen, F.; Wang, L. The Autoimmune Encephalitis-Related Cytokine TSLP in the Brain Primes Neuroinflammation by Activating the JAK2-NLRP3 Axis. Clin. Exp. Immunol. 2022, 207, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jian, Z.; Zhong, Y.; Ye, Y.; Zhang, Y.; Hu, X.; Pu, B.; Gu, L.; Xiong, X. Janus Kinase Inhibition Ameliorates Ischemic Stroke Injury and Neuroinflammation Through Reducing NLRP3 Inflammasome Activation via JAK2/STAT3 Pathway Inhibition. Front. Immunol. 2021, 12, 714943. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Shapiro, L.C.; de Oliveira, S.; Rivera-Pena, B.; Verma, A.; Shastri, A. Therapeutic Targeting of the Inflammasome in Myeloid Malignancies. Blood Cancer J. 2021, 11, 152. [Google Scholar] [CrossRef]
- di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef]
- Zhong, C.; Wang, R.; Hua, M.; Zhang, C.; Han, F.; Xu, M.; Yang, X.; Li, G.; Hu, X.; Sun, T.; et al. NLRP3 Inflammasome Promotes the Progression of Acute Myeloid Leukemia via IL-1β Pathway. Front. Immunol. 2021, 12, 2113. [Google Scholar] [CrossRef]
- Lu, F.; Zhao, Y.; Pang, Y.; Ji, M.; Sun, Y.; Wang, H.; Zou, J.; Wang, Y.; Li, G.; Sun, T.; et al. NLRP3 Inflammasome Upregulates PD-L1 Expression and Contributes to Immune Suppression in Lymphoma. Cancer Lett. 2021, 497, 178–189. [Google Scholar] [CrossRef]
- Koehn, B.H.; Saha, A.; McDonald-Hyman, C.; Loschi, M.; Thangavelu, G.; Ma, L.; Zaiken, M.; Dysthe, J.; Krepps, W.; Panthera, J.; et al. Danger-Associated Extracellular ATP Counters MDSC Therapeutic Efficacy in Acute GVHD. Blood 2019, 134, 1670–1682. [Google Scholar] [CrossRef]
- Hofbauer, D.; Mougiakakos, D.; Broggini, L.; Zaiss, M.; Büttner-Herold, M.; Bach, C.; Spriewald, B.; Neumann, F.; Bisht, S.; Nolting, J.; et al. Β2-Microglobulin Triggers NLRP3 Inflammasome Activation in Tumor-Associated Macrophages to Promote Multiple Myeloma Progression. Immunity 2021, 54, 1772–1787.e9. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Kamimura, S.; Arora, T.; Smith, M.L.; Almeida, L.E.F.; Combs, C.A.; Thein, S.L.; Quezado, Z.M.N. NLRP3 Inflammasome and Bruton Tyrosine Kinase Inhibition Interferes with Upregulated Platelet Aggregation and in Vitro Thrombus Formation in Sickle Cell Mice. Biochem. Biophys. Res. Commun. 2021, 555, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The Platelet NLRP3 Inflammasome Is Upregulated in Sickle Cell Disease via HMGB1/TLR4 and Bruton Tyrosine Kinase. Blood Adv. 2018, 2, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.N.R. Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s Tyrosine Kinase Is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; List, A. The Central Role of Inflammatory Signaling in the Pathogenesis of Myelodysplastic Syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parciante, E.; Cumbo, C.; Anelli, L.; Zagaria, A.; Redavid, I.; Minervini, A.; Conserva, M.R.; Tota, G.; Coccaro, N.; Tarantini, F.; et al. The Role of NLRP3, a Star of Excellence in Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2023, 24, 4860. https://doi.org/10.3390/ijms24054860
Parciante E, Cumbo C, Anelli L, Zagaria A, Redavid I, Minervini A, Conserva MR, Tota G, Coccaro N, Tarantini F, et al. The Role of NLRP3, a Star of Excellence in Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2023; 24(5):4860. https://doi.org/10.3390/ijms24054860
Chicago/Turabian StyleParciante, Elisa, Cosimo Cumbo, Luisa Anelli, Antonella Zagaria, Immacolata Redavid, Angela Minervini, Maria Rosa Conserva, Giuseppina Tota, Nicoletta Coccaro, Francesco Tarantini, and et al. 2023. "The Role of NLRP3, a Star of Excellence in Myeloproliferative Neoplasms" International Journal of Molecular Sciences 24, no. 5: 4860. https://doi.org/10.3390/ijms24054860