
Psoriatic Arthritis: Pathogenesis and Targeted Therapies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology
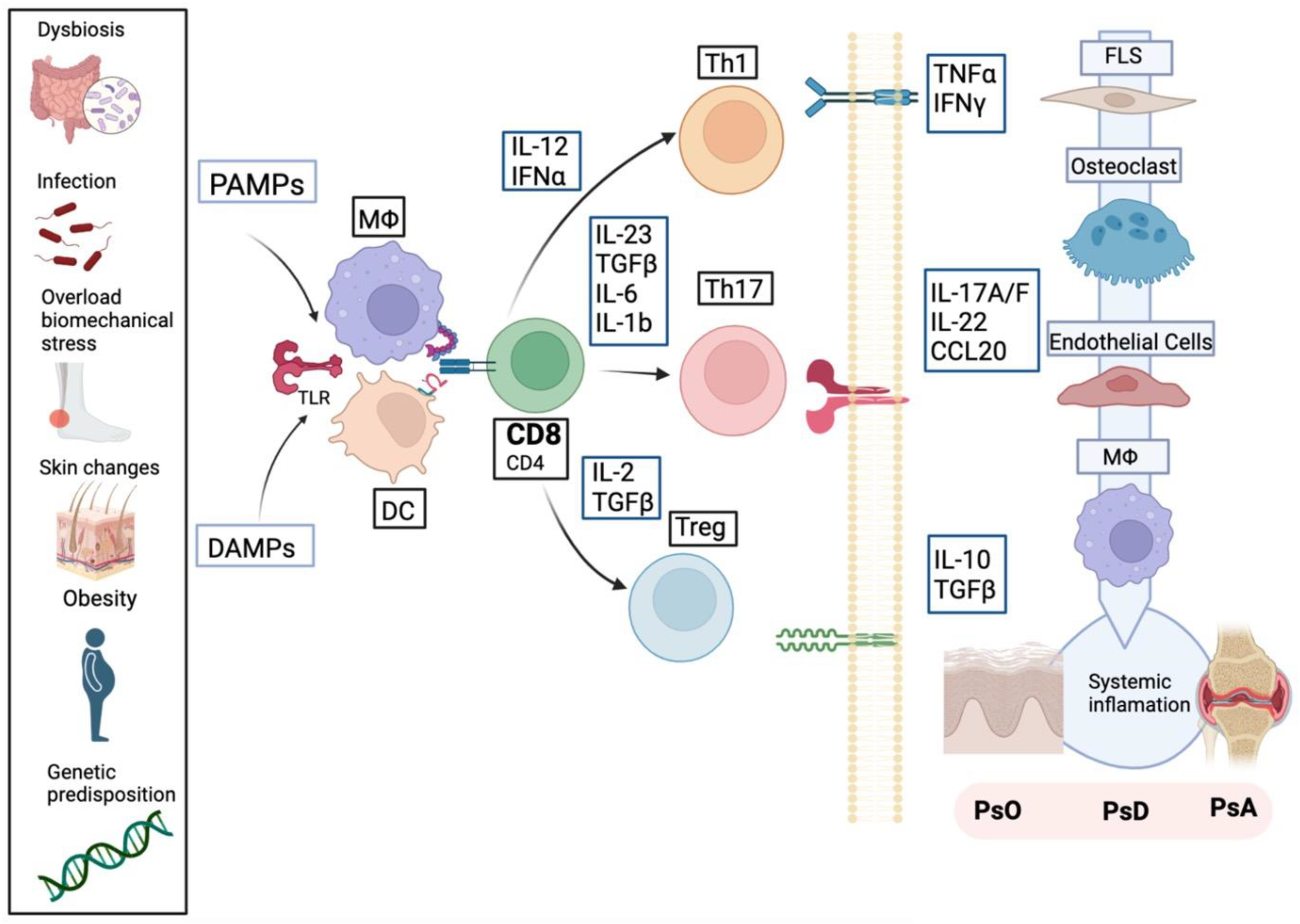
3. Pathogenesis
3.1. Genetic Susceptibility
3.2. Epigenetics/DNA Methylation
3.3. Microbiome and Dysbiosis
3.4. Biomechanical Stress
3.5. Obesity
3.6. Smoking
3.7. Infections
3.8. Innate and Adaptative Immune System: Pathological Processes in PsA
4. Psoriatic Synovitis
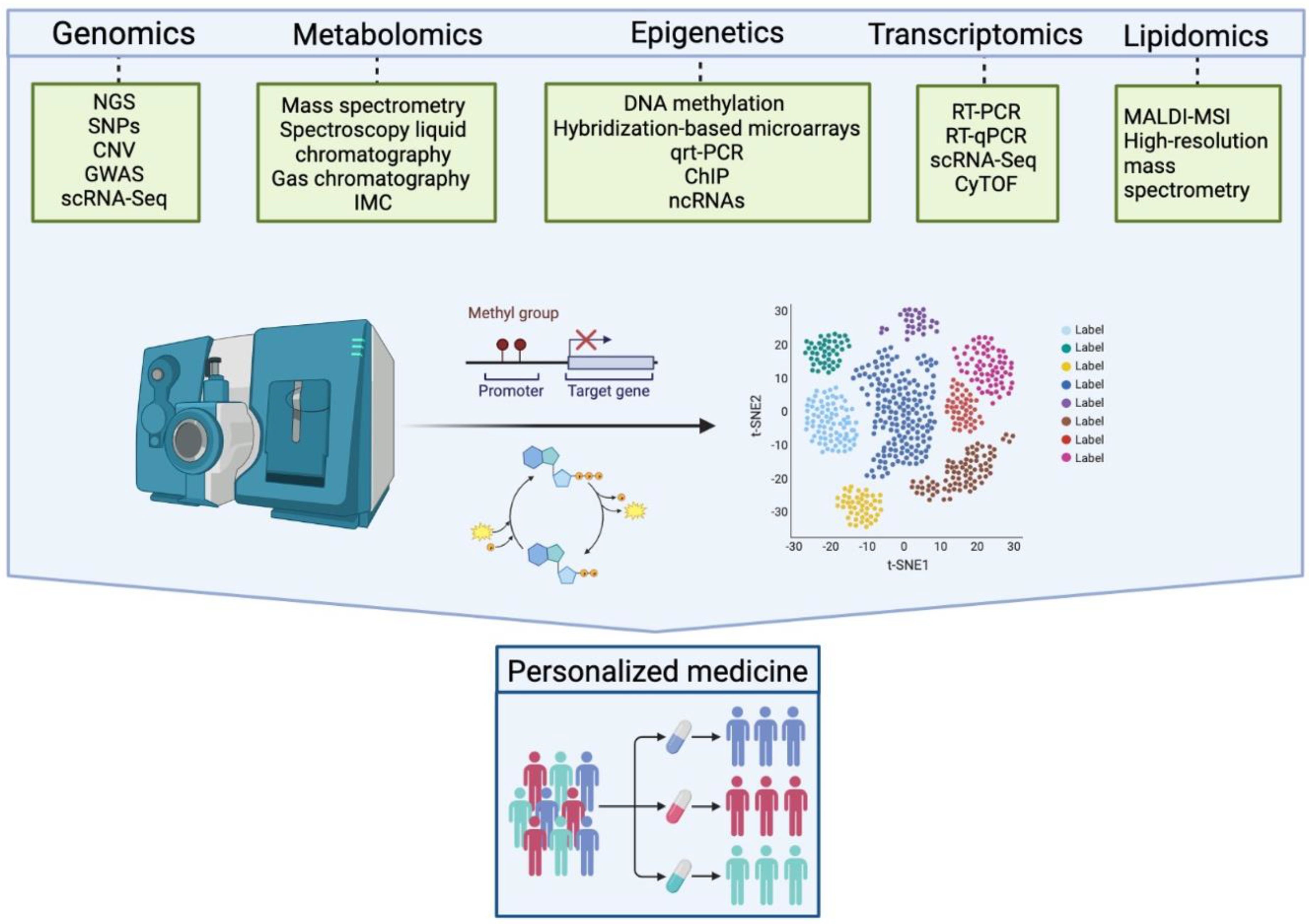
4.1. Single Cell Analysis and Other Omics Applied to Synovitis: A New Perspective on the Role of Synovial Fibroblasts
4.2. T-Cell Subtypes in PsA
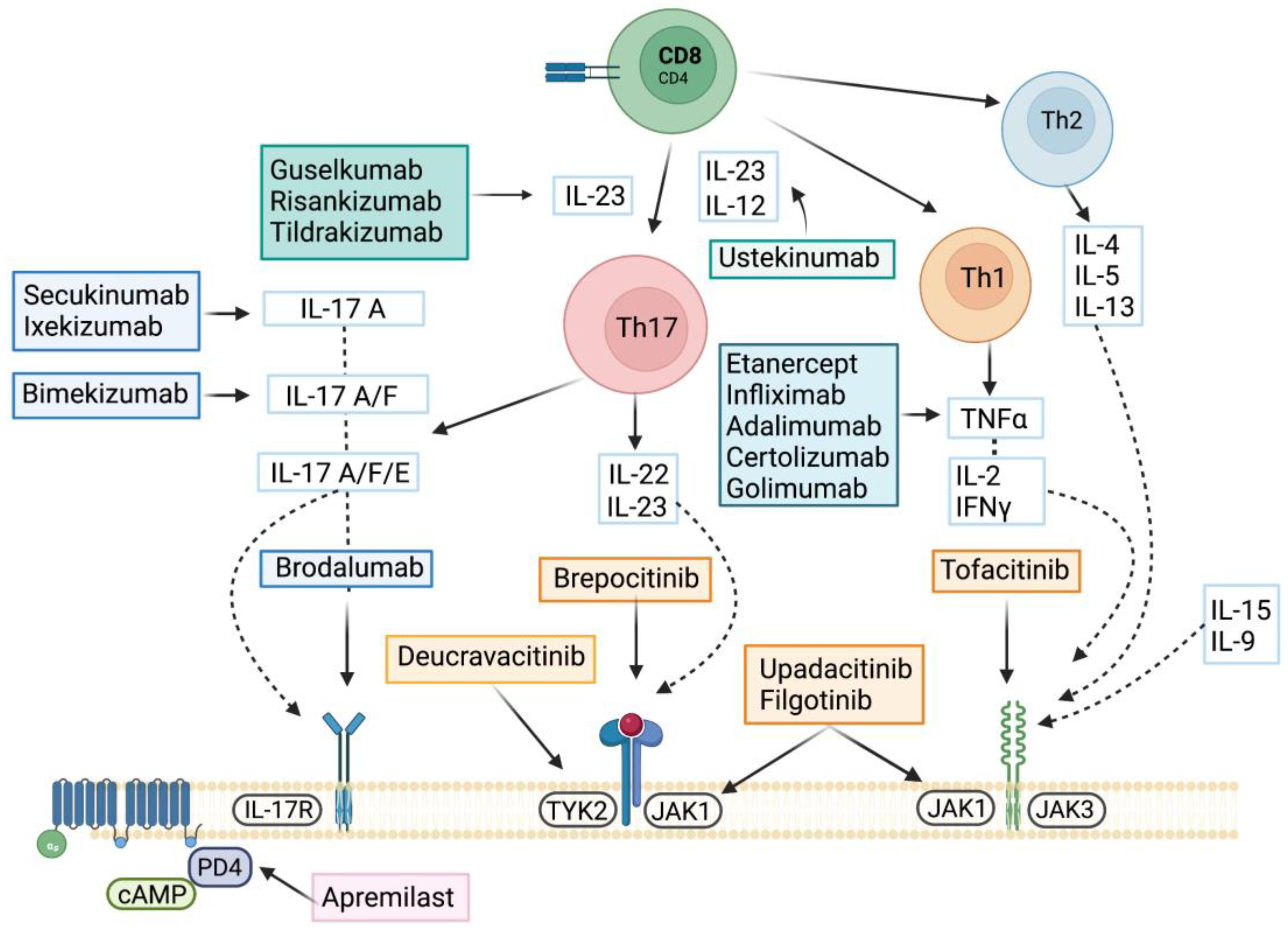
5. Targeted Therapies
5.1. Apremilast
5.2. TNF Inhibitors
5.3. IL-17/IL-23 Axis
5.3.1. IL23 Inhibitors
5.3.2. IL-17 Inhibitors
5.4. JAK Inhibitors
6. Future Perspectives in Psoriatic Arthritis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alinaghi, F.; Calov, M.; Kristensen, L.E.; Gladman, D.D.; Coates, L.C.; Jullien, D.; Gottlieb, A.B.; Gisondi, P.; Wu, J.J.; Thyssen, J.P.; et al. Prevalence of Psoriatic Arthritis in Patients with Psoriasis: A Systematic Review and Meta-Analysis of Observational and Clinical Studies. J. Am. Acad. Dermatol. 2019, 80, 251–265.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, E.L.; Orbai, A.M.; Ritchlin, C.T. Targeting Extra-Articular Manifestations in PsA: A Closer Look at Enthesitis and Dactylitis. Curr. Opin. Rheumatol. 2015, 27, 111–117. [Google Scholar] [CrossRef]
- Gossec, L.; Baraliakos, X.; Kerschbaumer, A.; de Wit, M.; McInnes, I.; Dougados, M.; Primdahl, J.; McGonagle, D.G.; Aletaha, D.; Balanescu, A.; et al. EULAR Recommendations for the Management of Psoriatic Arthritis with Pharmacological Therapies: 2019 Update. Ann. Rheum. Dis. 2020, 79, S700–S712. [Google Scholar] [CrossRef]
- Lubrano, E.; Scriffignano, S.; Azuaga, A.B.; Ramirez, J.; Cañete, J.D.; Perrotta, F.M. Impact of Comorbidities on Disease Activity, Patient Global Assessment, and Function in Psoriatic Arthritis: A Cross-Sectional Study. Rheumatol. Ther. 2020, 7, 825–836. [Google Scholar] [CrossRef]
- Perez-Chada, L.M.; Merola, J.F. Comorbidities Associated with Psoriatic Arthritis: Review and Update. Clin. Immunol. 2020, 214, 108397. [Google Scholar] [CrossRef] [PubMed]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-Wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. Am. J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef] [Green Version]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and Autoimmunity across Rheumatic and Musculoskeletal Diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595. [Google Scholar] [CrossRef]
- Julià, A.; Pinto, J.A.; Gratacós, J.; Queiró, R.; Ferrándiz, C.; Fonseca, E.; Montilla, C.; Torre-Alonso, J.C.; Puig, L.; Venegas, J.J.P.; et al. A Deletion at ADAMTS9-MAGI1 Locus Is Associated with Psoriatic Arthritis Risk. Ann. Rheum. Dis. 2015, 74, 1875–1881. [Google Scholar] [CrossRef] [Green Version]
- Belasco, J.; Louie, J.S.; Gulati, N.; Wei, N.; Nograles, K.; Fuentes-Duculan, J.; Mitsui, H.; Suárez-Fariñas, M.; Krueger, J.G. Comparative Genomic Profiling of Synovium versus Skin Lesions in Psoriatic Arthritis. Arthritis Rheumatol. 2015, 67, 934–944. [Google Scholar] [CrossRef]
- Anchang, C.G.; Xu, C.; Raimondo, M.G.; Atreya, R.; Maier, A.; Schett, G.; Zaburdaev, V.; Rauber, S.; Ramming, A. The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2021, 22, 7506. [Google Scholar] [CrossRef]
- Najm, A.; Goodyear, C.S.; McInnes, I.B.; Siebert, S. Phenotypic Heterogeneity in Psoriatic Arthritis: Towards Tissue Pathology-Based Therapy. Nat. Rev. Rheumatol. 2023, 19, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Gladman, D.D.; Mease, P.J.; Smith, N.; Margolis, D.J.; Nijsten, T.; Stern, R.S.; Feldman, S.R.; Rolstad, T. Epidemiology of Psoriatic Arthritis in the Population of the United States. J. Am. Acad. Dermatol. 2005, 53, 573.e1–573.e13. [Google Scholar] [CrossRef]
- Madland, T.M.; Apalset, E.M.; Johannessen, A.E.; Rossebö, B.; Brun, J.G. Prevalence, Disease Manifestations, and Treatment of Psoriatic Arthritis in Western Norway. J. Rheumatol. 2005, 32, 1918–1922. [Google Scholar] [PubMed]
- Hoff, M.; Gulati, A.M.; Romundstad, P.R.; Kavanaugh, A.; Haugeberg, G. Prevalence and Incidence Rates of Psoriatic Arthritis in Central Norway: Data from the Nord-Trøndelag Health Study (HUNT). Ann. Rheum. Dis. 2015, 74, 60–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hukuda, S.; Minami, M.; Saito, T.; Mitsui, H.; Matsui, N.; Komatsubara, Y.; Makino, H.; Shibata, T.; Shingu, M.; Sakou, T.; et al. Spondyloarthropathies in Japan: Nationwide Questionnaire Survey Performed by the Japan Ankylosing Spondylitis Society. J. Rheumatol. 2001, 28, 554–559. [Google Scholar]
- Li, R.; Sun, J.; Ren, L.-M.; Wang, H.-Y.; Liu, W.-H.; Zhang, X.-W.; Chen, S.; Mu, R.; He, J.; Zhao, Y.; et al. Epidemiology of Eight Common Rheumatic Diseases in China: A Large-Scale Cross-Sectional Survey in Beijing. Rheumatology 2012, 51, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and Incidence of Psoriatic Arthritis: A Systematic Review and Meta-Analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ohtsuki, M.; Sano, S.; Igarashi, A.; Morita, A.; Okuyama, R.; Kawada, A. Epidemiological Analysis of Psoriatic Arthritis Patients in Japan. J. Dermatol. 2016, 43, 1193–1196. [Google Scholar] [CrossRef]
- Karmacharya, P.; Crowson, C.S.; Bekele, D.; Achenbach, S.J.; Davis, J.M.; Ogdie, A.; Duarte-García, A.; Ernste, F.C.; Maradit-Kremers, H.; Tollefson, M.M.; et al. The Epidemiology of Psoriatic Arthritis over 5 Decades: A Population-Based Study. Arthritis Rheumatol. 2021, 73, 1878. [Google Scholar] [CrossRef] [PubMed]
- Eder, L.; Thavaneswaran, A.; Chandran, V.; Gladman, D.D. Gender Difference in Disease Expression, Radiographic Damage and Disability among Patients with Psoriatic Arthritis. Ann. Rheum. Dis. 2013, 72, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Kalyoncu, U.; Bayindir, Ö.; Öksüz, M.F.; Doğru, A.; Kimyon, G.; Tarhan, E.F.; Erden, A.; Yavuz, Ş.; Can, M.; Çetin, G.Y.; et al. The Psoriatic Arthritis Registry of Turkey: Results of a Multicentre Registry on 1081 Patients. Rheumatology 2017, 56, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duruöz, M.T.; Gezer, H.H.; Nas, K.; Kılıç, E.; Sargın, B.; Kasman, S.A.; Alkan, H.; Şahin, N.; Cengiz, G.; Cüzdan, N.; et al. Gender-Related Differences in Disease Activity and Clinical Features in Patients with Peripheral Psoriatic Arthritis: A Multi-Center Study. Jt. Bone Spine 2021, 88, 105177. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Triggianese, P.; Conigliaro, P.; Tonelli, M.; Gigliucci, G.; Novelli, L.; Teoli, M.; Perricone, R. A 2-Year Observational Study on Treatment Targets in Psoriatic Arthritis Patients Treated with TNF Inhibitors. Clin. Rheumatol. 2017, 36, 2253–2260. [Google Scholar] [CrossRef]
- Stober, C.; Ye, W.; Guruparan, T.; Htut, E.; Clunie, G.; Jadon, D. Prevalence and Predictors of Tumour Necrosis Factor Inhibitor Persistence in Psoriatic Arthritis. Rheumatology 2018, 57, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Navarini, L.; Costa, L.; Tasso, M.; Chimenti, M.S.; Currado, D.; Fonti, G.L.; Ciccozzi, M.; Margiotta, D.P.E.; Benigno, C.; de Martino, E.; et al. Retention Rates and Identification of Factors Associated with Anti-TNFα, Anti-IL17, and Anti-IL12/23R Agents Discontinuation in Psoriatic Arthritis Patients: Results from a Real-World Clinical Setting. Clin. Rheumatol. 2020, 39, 2663–2670. [Google Scholar] [CrossRef]
- Favalli, E.G.; Conti, F.; Selmi, C.; Iannone, F.; Bucci, R.; D’Onofrio, F.; Carlino, G.; Santo, L.; Semeraro, A.; Zuccaro, C.; et al. Retrospective Evaluation of Patient Profiling and Effectiveness of Apremilast in an Italian Multicentric Cohort of Psoriatic Arthritis Patients. Clin. Exp. Rheumatol. 2020, 38, 19–26. [Google Scholar]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H.; Schölmerich, J.; Fleck, M.; Härle, P. Serum BAFF Strongly Correlates with PsA Activity in Male Patients Only—Is There a Role for Sex Hormones? Clin. Exp. Rheumatol. 2010, 28, 813–819. [Google Scholar]
- Wilson, F.C.; Icen, M.; Crowson, C.S.; McEvoy, M.T.; Gabriel, S.E.; Kremers, H.M. Time Trends in Epidemiology and Characteristics of Psoriatic Arthritis Over 3 Decades: A Population-Based Study. J. Rheumatol. 2009, 36, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Polachek, A.; Al-Johani, R.; Li, S.; Ye, J.Y.; Chandran, V.; Gladman, D. Late Onset Psoriatic Arthritis in a Longitudinal Cohort: Disease Presentation, Activity over Time and Prognosis. Semin. Arthritis Rheum. 2019, 48, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Punzi, L.; Pianon, M.; Rossini, P.; Schiavon, F.; Gambari, P.F. Clinical and Laboratory Manifestations of Elderly Onset Psoriatic Arthritis: A Comparison with Younger Onset Disease. Ann. Rheum. Dis. 1999, 58, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobak, S.; Yildiz, F.; Karaarslan, A.; Semiz, H.; Orman, M. Characteristics of Turkish Patients with Elderly Onset Psoriatic Arthritis: A Retrospective Cohort Study. Medicine 2017, 96, e7833. [Google Scholar] [CrossRef]
- Bilgin, E.; Aydin, S.Z.; Tinazzi, I.; Bayindir, Ö.; Kimyon, G.; Özişler, C.; Doğru, A.; Dalkiliç, E.; Aksu, K.; Çetin, G.Y.; et al. Disease Characteristics of Psoriatic Arthritis Patients May Differ According to Age at Psoriasis Onset: Cross-Sectional Data from the Psoriatic Arthritis-International Database. Clin. Exp. Rheumatol. 2021, 39, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Rahman, P.; Ritchlin, C.; McInnes, I.B.; Elewaut, D.; Scher, J.U. Psoriatic Arthritis from a Mechanistic Perspective. Nat. Rev. Rheumatol. 2022, 18, 311–325. [Google Scholar] [CrossRef]
- Gladman, D.D.; Farewell, V.T.; Pellett, F.; Schentag, C.; Rahman, P. HLA Is a Candidate Region for Psoriatic Arthritis: Evidence for Excessive HLA Sharing in Sibling Pairs. Hum. Immunol. 2003, 64, 887–889. [Google Scholar] [CrossRef]
- Chandran, V.; Schentag, C.T.; Brockbank, J.E.; Pellett, F.J.; Shanmugarajah, S.; Toloza, S.M.A.; Rahman, P.; Gladman, D.D. Familial Aggregation of Psoriatic Arthritis. Ann. Rheum. Dis. 2009, 68, 664–667. [Google Scholar] [CrossRef]
- Li, Q.; Chandran, V.; Tsoi, L.; O’Rielly, D.; Nair, R.P.; Gladman, D.; Elder, J.T.; Rahman, P. Quantifying Differences in Heritability among Psoriatic Arthritis (PsA), Cutaneous Psoriasis (PsC) and Psoriasis Vulgaris (PsV). Sci. Rep. 2020, 10, 4925. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, O.; Haroon, M.; Giles, J.T.; Winchester, R. Concepts of Pathogenesis in Psoriatic Arthritis: Genotype Determines Clinical Phenotype. Arthritis Res. Ther. 2015, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Han, B.; Tsoi, L.C.; Stuart, P.E.; Ellinghaus, E.; Tejasvi, T.; Chandran, V.; Pellett, F.; Pollock, R.; Bowcock, A.M.; et al. Fine Mapping Major Histocompatibility Complex Associations in Psoriasis and Its Clinical Subtypes. Am. J. Hum. Genet. 2014, 95, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowes, J.; Budu-Aggrey, A.; Huffmeier, U.; Uebe, S.; Steel, K.; Hebert, H.L.; Wallace, C.; Massey, J.; Bruce, I.N.; Bluett, J.; et al. Dense Genotyping of Immune-Related Susceptibility Loci Reveals New Insights into the Genetics of Psoriatic Arthritis. Nat. Commun. 2015, 6, 6046. [Google Scholar] [CrossRef] [Green Version]
- Patrick, M.T.; Stuart, P.E.; Raja, K.; Chi, S.; He, Z.; Voorhees, J.J.; Tejasvi, T.; Gudjonsson, J.E.; Michelle Kahlenberg, J.; Chandran, V.; et al. Integrative Approach to Reveal Cell Type Specificity and Gene Candidates for Psoriatic Arthritis Outside the MHC. Front. Genet. 2019, 10, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ubreva, J.; de La Calle-Fabregat, C.; Li, T.; Ciudad, L.; Ballestar, M.L.; Català-Moll, F.; Morante-Palacios, O.; Garcia-Gomez, A.; Celis, R.; Humby, F.; et al. Inflammatory Cytokines Shape a Changing DNA Methylome in Monocytes Mirroring Disease Activity in Rheumatoid Arthritis. Ann. Rheum. Dis. 2019, 78, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- de la Calle-Fabregat, C.; Niemantsverdriet, E.; Cañete, J.D.; Li, T.; van der Helm-van Mil, A.H.M.; Rodríguez-Ubreva, J.; Ballestar, E. Prediction of the Progression of Undifferentiated Arthritis to Rheumatoid Arthritis Using DNA Methylation Profiling. Arthritis Rheumatol. 2021, 73, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- de la Calle-Fabregat, C.; Rodríguez-Ubreva, J.; Ciudad, L.; Ramírez, J.; Celis, R.; Azuaga, A.B.; Cuervo, A.; Graell, E.; Pérez-García, C.; Díaz-Torné, C.; et al. The Synovial and Blood Monocyte DNA Methylomes Mirror Prognosis, Evolution, and Treatment in Early Arthritis. JCI Insight 2022, 7, e158783. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pitzalis, C. Progress Continues in Prediction of the Response to Treatment of RA. Nat. Rev. Rheumatol. 2023, 19, 68–69. [Google Scholar] [CrossRef]
- Deng, M.; Su, Y.; Wu, R.; Li, S.; Zhu, Y.; Tang, G.; Shi, X.; Zhou, T.; Zhao, M.; Lu, Q. DNA Methylation Markers in Peripheral Blood for Psoriatic Arthritis. J. Dermatol. Sci. 2022, 108, 39–47. [Google Scholar] [CrossRef]
- Charras, A.; Garau, J.; Hofmann, S.R.; Carlsson, E.; Cereda, C.; Russ, S.; Abraham, S.; Hedrich, C.M. DNA Methylation Patterns in CD8+ T Cells Discern Psoriasis From Psoriatic Arthritis and Correlate With Cutaneous Disease Activity. Front. Cell Dev. Biol. 2021, 9, 746145. [Google Scholar] [CrossRef]
- Burgueño, J.F.; Abreu, M.T. Epithelial Toll-like Receptors and Their Role in Gut Homeostasis and Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 263–278. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Gracey, E.; Vereecke, L.; McGovern, D.; Fröhling, M.; Schett, G.; Danese, S.; de Vos, M.; van den Bosch, F.; Elewaut, D. Revisiting the Gut-Joint Axis: Links between Gut Inflammation and Spondyloarthritis. Nat. Rev. Rheumatol. 2020, 16, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Alessandro, R.; Luchetti, M.M.; Milling, S.; Saieva, L.; Cypers, H.; Stampone, T.; di Benedetto, P.; et al. Dysbiosis and Zonulin Upregulation Alter Gut Epithelial and Vascular Barriers in Patients with Ankylosing Spondylitis. Ann. Rheum. Dis. 2017, 76, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Boix-Amorós, A.; Badri, M.H.; Manasson, J.; Blank, R.B.; Haberman, R.H.; Neimann, A.L.; Girija, P.V.; Jimenez Hernandez, A.; Heguy, A.; Koralov, S.; et al. Alterations in the Cutaneous Microbiome of Patients with Psoriasis and Psoriatic Arthritis Reveal Similarities between Non-Lesional and Lesional Skin. Ann. Rheum. Dis. 2022. online first. [Google Scholar] [CrossRef]
- Gracey, E.; Burssens, A.; Cambré, I.; Schett, G.; Lories, R.; McInnes, I.B.; Asahara, H.; Elewaut, D. Tendon and Ligament Mechanical Loading in the Pathogenesis of Inflammatory Arthritis. Nat. Rev. Rheumatol. 2020, 16, 193–207. [Google Scholar] [CrossRef]
- Cambré, I.; Gaublomme, D.; Burssens, A.; Jacques, P.; Schryvers, N.; de Muynck, A.; Meuris, L.; Lambrecht, S.; Carter, S.; de Bleser, P.; et al. Mechanical Strain Determines the Site-Specific Localization of Inflammation and Tissue Damage in Arthritis. Nat. Commun. 2018, 9, 4613. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; Gibbon, W.; Emery, P. Classification of Inflammatory Arthritis by Enthesitis. Lancet 1998, 352, 1137–1140. [Google Scholar] [CrossRef]
- Bridgewood, C.; Watad, A.; Russell, T.; Palmer, T.M.; Marzo-Ortega, H.; Khan, A.; Millner, P.A.; Dunsmuir, R.; Rao, A.; Loughenbury, P.; et al. Identification of Myeloid Cells in the Human Enthesis as the Main Source of Local IL-23 Production. Ann. Rheum. Dis. 2019, 78, 929–933. [Google Scholar] [CrossRef] [Green Version]
- Thorarensen, S.M.; Lu, N.; Ogdie, A.; Gelfand, J.M.; Choi, H.K.; Love, T.J. Physical Trauma Recorded in Primary Care Is Associated with the Onset of Psoriatic Arthritis among Patients with Psoriasis. Ann. Rheum. Dis. 2017, 76, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Bhole, V.M.; Choi, H.K.; Burns, L.C.; Kellet, C.V.; Lacaille, D.V.; Gladman, D.D.; Dutz, J.P. Differences in Body Mass Index among Individuals with PsA, Psoriasis, RA and the General Population. Rheumatology 2012, 51, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, D.; Snekvik, I.; Schlesinger, S.; Norat, T.; Riboli, E.; Vatten, L.J. Body Mass Index, Abdominal Fatness, Weight Gain and the Risk of Psoriasis: A Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Eur. J. Epidemiol. 2018, 33, 1163–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russolillo, A.; Iervolino, S.; Peluso, R.; Lupoli, R.; di Minno, A.; Pappone, N.; di Minno, M.N.D. Obesity and Psoriatic Arthritis: From Pathogenesis to Clinical Outcome and Management. Rheumatology 2013, 52, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Facciorusso, A.; Singh, A.G.; van de Casteele, N.; Zarrinpar, A.; Prokop, L.J.; Grunvald, E.L.; Curtis, J.R.; Sandborn, W.J. Obesity and Response to Anti-Tumor Necrosis Factor-α Agents in Patients with Select Immune-Mediated Inflammatory Diseases: A Systematic Review and Meta-Analysis. PLoS ONE 2018, 13, e0195123. [Google Scholar] [CrossRef] [Green Version]
- Gazel, U.; Ayan, G.; Solmaz, D.; Akar, S.; Aydin, S.Z. The Impact of Smoking on Prevalence of Psoriasis and Psoriatic Arthritis. Rheumatology 2020, 59, 2695–2710. [Google Scholar] [CrossRef]
- Nguyen, U.S.D.T.; Zhang, Y.; Lu, N.; Louie-Gao, Q.; Niu, J.; Ogdie, A.; Gelfand, J.M.; LaValley, M.P.; Dubreuil, M.; Sparks, J.A.; et al. Smoking Paradox in the Development of Psoriatic Arthritis among Patients with Psoriasis: A Population-Based Study. Ann. Rheum. Dis. 2018, 77, 119–123. [Google Scholar] [CrossRef]
- Ladehesa-Pineda, M.L.; Ortega-Castro, R.; Puche-Larrubia, M.Á.; Granados, R.E.M.; Dougados, M.; Collantes-Estévez, E.; López-Medina, C. Smoking and Alcohol Consumption Are Associated with Peripheral Musculoskeletal Involvement in Patients with Spondyloarthritis (Including Psoriatic Arthritis). Results from the ASAS-PerSpA Study. Semin. Arthritis Rheum. 2023, 58, 152146. [Google Scholar] [CrossRef]
- Telfer, N.R.; Chalmers, R.J.G.; Whale, K.; Colman, G. The Role of Streptococcal Infection in the Initiation of Guttate Psoriasis. Arch. Dermatol. 1992, 128, 39–42. [Google Scholar] [CrossRef]
- Wang, Q.; Vasey, F.B.; Mahfood, J.P.; Valeriano, J.; Kanik, K.S.; Anderson, B.E.; Bridgeford, P.H. V2 regions of 16s ribosomal rna used as a molecular marker for the species identification of streptococci in peripheral blood and synovial fluid from patients with psoriatic arthritis. Arthritis Rheum. 1999, 42, 2055–2059. [Google Scholar] [CrossRef]
- Thrastardottir, T.; Meer, E.; Hauksdottir, A.; Gudbjornsson, B.; Kristinsson, S.Y.; Ogdie, A.; Love, T.J. Strong Site-Specific Association of Pharyngeal Cultures with the Onset of Psoriatic Arthritis and Psoriasis, Regardless of Pathogen. Rheumatology 2023, 62, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Candia, L.; Marquez, J.; Hernandez, C.; Zea, A.H.; Espinoza, L.R. Toll-like Receptor-2 Expression Is Upregulated in Antigen-Presenting Cells from Patients with Psoriatic Arthritis: A Pathogenic Role for Innate Immunity? J. Rheumatol. 2007, 34, 374–379. [Google Scholar] [PubMed]
- Davila, E.; Kolls, J. A “Toll” for Th17 Cell Expansion. J. Leukoc. Biol. 2010, 88, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Soare, A.; Weber, S.; Maul, L.; Rauber, S.; Gheorghiu, A.M.; Luber, M.; Houssni, I.; Kleyer, A.; von Pickardt, G.; Gado, M.; et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J. Immunol. 2018, 200, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Diani, M.; Casciano, F.; Marongiu, L.; Longhi, M.; Altomare, A.; Pigatto, P.D.; Secchiero, P.; Gambari, R.; Banfi, G.; Manfredi, A.A.; et al. Increased Frequency of Activated CD8+ T Cell Effectors in Patients with Psoriatic Arthritis. Sci. Rep. 2019, 9, 10870. [Google Scholar] [CrossRef] [Green Version]
- Menon, B.; Gullick, N.J.; Walter, G.J.; Rajasekhar, M.; Garrood, T.; Evans, H.G.; Taams, L.S.; Kirkham, B.W. Interleukin-17+CD8+ T Cells Are Enriched in the Joints of Patients with Psoriatic Arthritis and Correlate with Disease Activity and Joint Damage Progression. Arthritis Rheumatol. 2014, 66, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Morishima, N.; Mizoguchi, I.; Takeda, K.; Mizuguchi, J.; Yoshimoto, T. TGF-Beta Is Necessary for Induction of IL-23R and Th17 Differentiation by IL-6 and IL-23. Biochem. Biophys. Res. Commun. 2009, 386, 105–110. [Google Scholar] [CrossRef]
- Veale, D.J.; Fearon, U. The Pathogenesis of Psoriatic Arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Ruiz de Morales, J.M.G.; Puig, L.; Daudén, E.; Cañete, J.D.; Pablos, J.L.; Martín, A.O.; Juanatey, C.G.; Adán, A.; Montalbán, X.; Borruel, N.; et al. Critical Role of Interleukin (IL)-17 in Inflammatory and Immune Disorders: An Updated Review of the Evidence Focusing in Controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef]
- Cañete, J.D.; Pablos, J.L.; Sanmartí, R.; Mallofré, C.; Marsal, S.; Maymó, J.; Gratacós, J.; Mezquita, J.; Mezquita, C.; Cid, M.C. Antiangiogenic Effects of Anti-Tumor Necrosis Factor Alpha Therapy with Infliximab in Psoriatic Arthritis. Arthritis Rheum. 2004, 50, 1636–1641. [Google Scholar] [CrossRef]
- Suzuki, E.; Mellins, E.D.; Gershwin, M.E.; Nestle, F.O.; Adamopoulos, I.E. The IL-23/IL-17 Axis in Psoriatic Arthritis. Autoimmun. Rev. 2014, 13, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 Induces Spondyloarthropathy by Acting on ROR-Γt+ CD3+CD4-CD8- Entheseal Resident T Cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 Cytokines Interleukin (IL)-17 and IL-22 Modulate Distinct Inflammatory and Keratinocyte-Response Pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vičić, M.; Kaštelan, M.; Brajac, I.; Sotošek, V.; Massari, L.P. Current Concepts of Psoriasis Immunopathogenesis. Int. J. Mol. Sci. 2021, 22, 11574. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, M.G.; Baeten, D.L. Immunopathology of Synovitis: From Histology to Molecular Pathways. Rheumatology 2016, 55, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Veale, D.; Yanni, G.; Rogers, S.; Barnes, L.; Bresnihan, B.; Fitzgerald, O. Reduced Synovial Membrane Macrophage Numbers, ELAM-1 Expression, and Lining Layer Hyperplasia in Psoriatic Arthritis as Compared with Rheumatoid Arthritis. Arthritis Rheum. 1993, 36, 893–900. [Google Scholar] [CrossRef]
- Izquierdo, E.; Cañete, J.D.; Celis, R.; del Rey, M.J.; Usategui, A.; Marsal, S.; Sanmartí, R.; Criado, G.; Pablos, J.L. Synovial Fibroblast Hyperplasia in Rheumatoid Arthritis: Clinicopathologic Correlations and Partial Reversal by Anti-Tumor Necrosis Factor Therapy. Arthritis Rheum. 2011, 63, 2575–2583. [Google Scholar] [CrossRef]
- Cuervo, A.; Celis, R.; Julià, A.; Usategui, A.; Faré, R.; Ramírez, J.; Azuaga, A.B.; Lorenzo, A.; Sanmartí, R.; Pablos, J.L.; et al. Synovial Immunohistological Biomarkers of the Classification of Undifferentiated Arthritis Evolving to Rheumatoid or Psoriatic Arthritis. Front. Med. 2021, 8, 656667. [Google Scholar] [CrossRef]
- Gao, W.; McGarry, T.; Orr, C.; McCormick, J.; Veale, D.J.; Fearon, U. Tofacitinib Regulates Synovial Inflammation in Psoriatic Arthritis, Inhibiting STAT Activation and Induction of Negative Feedback Inhibitors. Ann. Rheum. Dis. 2016, 75, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Kruithof, E.; de Rycke, L.; Vandooren, B.; Wyns, B.; Boullart, L.; Hoffman, I.E.A.; Boots, A.M.; Veys, E.M.; de Keyser, F. Diagnostic Classification of Spondylarthropathy and Rheumatoid Arthritis by Synovial Histopathology: A Prospective Study in 154 Consecutive Patients. Arthritis Rheum. 2004, 50, 2931–2941. [Google Scholar] [CrossRef]
- Baeten, D.; Demetter, P.; Cuvelier, C.A.; Kruithof, E.; van Damme, N.; de Vos, M.; Veys, E.M.; de Keyser, F. Macrophages Expressing the Scavenger Receptor CD163: A Link between Immune Alterations of the Gut and Synovial Inflammation in Spondyloarthropathy. J. Pathol. 2002, 196, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador, G.; Sanmarti, R.; Garcia-Peiró, A.; Rodríguez-Cros, J.R.; Muñoz-Gómez, J.; Cañete, J.D. P53 Expression in Rheumatoid and Psoriatic Arthritis Synovial Tissue and Association with Joint Damage. Ann. Rheum. Dis. 2005, 64, 183–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentelsaz-Romero, S.; Cuervo, A.; Estrada-Capetillo, L.; Celis, R.; García-Campos, R.; Ramírez, J.; Sastre, S.; Samaniego, R.; Puig-Kröger, A.; Cañete, J.D. GM-CSF Expression and Macrophage Polarization in Joints of Undifferentiated Arthritis Patients Evolving to Rheumatoid Arthritis or Psoriatic Arthritis. Front. Immunol. 2021, 11, 613975. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Canavan, M.; Floudas, A.; Flynn, K.; Mullan, R.; Veale, D.J.; Fearon, U. CD209/CD14+ Dendritic Cells Characterization in Rheumatoid and Psoriatic Arthritis Patients: Activation, Synovial Infiltration, and Therapeutic Targeting. Front. Immunol. 2022, 12, 722349. [Google Scholar] [CrossRef]
- Cañete, J.D.; Santiago, B.; Cantaert, T.; Sanmartí, R.; Palacin, A.; Celis, R.; Graell, E.; Gil-Torregrosa, B.; Baeten, D.; Pablos, J.L. Ectopic Lymphoid Neogenesis in Psoriatic Arthritis. Ann. Rheum. Dis. 2007, 66, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Qiu, J.; Lin, Z.T.; Li, W.; Haley, C.; Mui, U.N.; Ning, J.; Tyring, S.K.; Wu, T. Identification of Novel Autoantibodies Associated With Psoriatic Arthritis. Arthritis Rheumatol. 2019, 71, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Frasca, L.; Palazzo, R.; Chimenti, M.S.; Alivernini, S.; Tolusso, B.; Bui, L.; Botti, E.; Giunta, A.; Bianchi, L.; Petricca, L.; et al. Anti-LL37 Antibodies Are Present in Psoriatic Arthritis (PsA) Patients: New Biomarkers in PsA. Front. Immunol. 2018, 9, 1936. [Google Scholar] [CrossRef] [Green Version]
- Koussiouris, J.; Chandran, V. Autoantibodies in Psoriatic Disease. J. Appl. Lab. Med. 2022, 7, 281–293. [Google Scholar] [CrossRef]
- Armas-González, E.; Díaz-Martín, A.; Domínguez-Luis, M.J.; Arce-Franco, M.T.; Herrera-García, A.; Hernández-Hernández, M.V.; Bustabad, S.; Usategui, A.; Pablos, J.L.; Cañete, J.D.; et al. Differential Antigen-Presenting B Cell Phenotypes from Synovial Microenvironment of Patients with Rheumatoid and Psoriatic Arthritis. J. Rheumatol. 2015, 42, 1825–1834. [Google Scholar] [CrossRef]
- Mavropoulos, A.; Zafiriou, E.; Simopoulou, T.; Brotis, A.G.; Liaskos, C.; Roussaki-Schulze, A.; Katsiari, C.G.; Bogdanos, D.P.; Sakkas, L.I. Apremilast Increases IL-10-Producing Regulatory B Cells and Decreases Proinflammatory T Cells and Innate Cells in Psoriatic Arthritis and Psoriasis. Rheumatology 2019, 58, 2240–2250. [Google Scholar] [CrossRef]
- Kruithof, E.; Baeten, D.; de Rycke, L.; Vandooren, B.; Foell, D.; Roth, J.; Cañete, J.D.; Boots, A.M.; Veys, E.M.; Keyser, F. de Synovial Histopathology of Psoriatic Arthritis, Both Oligo- and Polyarticular, Resembles Spondyloarthropathy More than It Does Rheumatoid Arthritis. Arthritis Res. Ther. 2005, 7, R569. [Google Scholar] [CrossRef] [Green Version]
- Fraser, A.; Fearon, U.; Billinghurst, R.C.; Ionescu, M.; Reece, R.; Barwick, T.; Emery, P.; Poole, A.R.; Veale, D.J. Turnover of Type II Collagen and Aggrecan in Cartilage Matrix at the Onset of Inflammatory Arthritis in Humans: Relationship to Mediators of Systemic and Local Inflammation. Arthritis Rheum. 2003, 48, 3085–3095. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally Distinct Disease-Associated Fibroblast Subsets in Rheumatoid Arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Floudas, A.; Smith, C.M.; Tynan, O.; Neto, N.; Krishna, V.; Wade, S.M.; Hanlon, M.; Cunningham, C.; Marzaioli, V.; Canavan, M.; et al. Distinct Stromal and Immune Cell Interactions Shape the Pathogenesis of Rheumatoid and Psoriatic Arthritis. Ann. Rheum. Dis. 2022, 81, 1224–1242. [Google Scholar] [CrossRef]
- Rauber, S.; Mohammadian, H.; Schmidkonz, C.; Atzinger, A.; Soare, A.; Maschauer, S.; Treutlein, C.; Angeli, M.; Raimondo, M.G.; Xu, C.; et al. Molecular Imaging with Fibroblast Activation Protein Tracers Depicts Inflammatory Joint Damage and Its Transition to Resolution of Inflammation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Penkava, F.; Velasco-Herrera, M.D.C.; Young, M.D.; Yager, N.; Nwosu, L.N.; Pratt, A.G.; Lara, A.L.; Guzzo, C.; Maroof, A.; Mamanova, L.; et al. Single-Cell Sequencing Reveals Clonal Expansions of pro-Inflammatory Synovial CD8 T Cells Expressing Tissue-Homing Receptors in Psoriatic Arthritis. Nat. Commun. 2020, 11, 4767. [Google Scholar] [CrossRef]
- Wade, S.M.; Canavan, M.; McGarry, T.; Low, C.; Wade, S.C.; Mullan, R.H.; Veale, D.J.; Fearon, U. Association of Synovial Tissue Polyfunctional T-Cells with DAPSA in Psoriatic Arthritis. Ann. Rheum. Dis. 2019, 78, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Coates, L.C.; Soriano, E.R.; Corp, N.; Bertheussen, H.; Callis Duffin, K.; Campanholo, C.B.; Chau, J.; Eder, L.; Fernández-Ávila, D.G.; FitzGerald, O.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): Updated Treatment Recommendations for Psoriatic Arthritis 2021. Nat. Rev. Rheumatol. 2022, 18, 465–479. [Google Scholar] [CrossRef]
- Schafer, P. Apremilast Mechanism of Action and Application to Psoriasis and Psoriatic Arthritis. Biochem. Pharmacol. 2012, 83, 1583–1590. [Google Scholar] [CrossRef]
- Claveau, D.; Chen, S.L.; O’Keefe, S.; Zaller, D.M.; Styhler, A.; Liu, S.; Huang, Z.; Nicholson, D.W.; Mancini, J.A. Preferential Inhibition of T Helper 1, but Not T Helper 2, Cytokines in Vitro by L-826,141 [4-{2-(3,4-Bisdifluromethoxyphenyl)-2-{4-(1,1,1,3,3,3-Hexafluoro-2-Hydroxypropan-2-Yl)-Phenyl]-Ethyl}-3-Methylpyridine-1-Oxide], a Potent and Selective Phosphodiesterase 4 Inhibitor. J. Pharmacol. Exp. Ther. 2004, 310, 752–760. [Google Scholar] [CrossRef]
- Eigler, A.; Siegmund, B.; Emmerich, U.; Baumann, K.H.; Hartmann, G.; Endres, S. Anti-Inflammatory Activities of CAMP-Elevating Agents: Enhancement of IL-10 Synthesis and Concurrent Suppression of TNF Production. J. Leukoc. Biol. 1998, 63, 101–107. [Google Scholar] [CrossRef]
- Papp, K.; Reich, K.; Leonardi, C.L.; Kircik, L.; Chimenti, S.; Langley, R.G.B.; Hu, C.C.; Stevens, R.M.; Day, R.M.; Gordon, K.B.; et al. Apremilast, an Oral Phosphodiesterase 4 (PDE4) Inhibitor, in Patients with Moderate to Severe Plaque Psoriasis: Results of a Phase III, Randomized, Controlled Trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J. Am. Acad. Dermatol. 2015, 73, 37–49. [Google Scholar] [CrossRef]
- Paul, C.; Cather, J.; Gooderham, M.; Poulin, Y.; Mrowietz, U.; Ferrandiz, C.; Crowley, J.; Hu, C.; Stevens, R.M.; Shah, K.; et al. Efficacy and Safety of Apremilast, an Oral Phosphodiesterase 4 Inhibitor, in Patients with Moderate-to-severe Plaque Psoriasis over 52 Weeks: A Phase III, Randomized Controlled Trial (ESTEEM 2). Br. J. Dermatol. 2015, 173, 1387–1399. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, A.; Mease, P.J.; Gomez-Reino, J.J.; Adebajo, A.O.; Wollenhaupt, J.; Gladman, D.D.; Lespessailles, E.; Hall, S.; Hochfeld, M.; Hu, C.; et al. Extended Report: Treatment of Psoriatic Arthritis in a Phase 3 Randomised, Placebo-Controlled Trial with Apremilast, an Oral Phosphodiesterase 4 Inhibitor. Ann. Rheum. Dis. 2014, 73, 1020. [Google Scholar] [CrossRef]
- Cutolo, M.; Myerson, G.E.; Fleischmann, R.M.; Liote, F.; Diaz-Gonzalez, F.; van den Bosch, F.; Marzo-Ortega, H.; Feist, E.; Shah, K.; Hu, C.; et al. A Phase III, Randomized, Controlled Trial of Apremilast in Patients with Psoriatic Arthritis: Results of the PALACE 2 Trial. J. Rheumatol. 2016, 43, 1724–1734. [Google Scholar] [CrossRef]
- Edwards, C.J.; Blanco, F.J.; Crowley, J.; Birbara, C.A.; Jaworski, J.; Aelion, J.; Stevens, R.M.; Vessey, A.; Zhan, X.; Bird, P. Extended Report: Apremilast, an Oral Phosphodiesterase 4 Inhibitor, in Patients with Psoriatic Arthritis and Current Skin Involvement: A Phase III, Randomised, Controlled Trial (PALACE 3). Ann. Rheum. Dis. 2016, 75, 1065. [Google Scholar] [CrossRef]
- Wells, A.F.; Edwards, C.J.; Kivitz, A.J.; Bird, P.; Nguyen, D.; Paris, M.; Teng, L.; Aelion, J.A. Apremilast Monotherapy in DMARD-Naive Psoriatic Arthritis Patients: Results of the Randomized, Placebo-Controlled PALACE 4 Trial. Rheumatology 2018, 57, 1253. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.J.; Fleischmann, R.; Deodhar, A.A.; Wollenhaupt, J.; Khraishi, M.; Kielar, D.; Woltering, F.; Stach, C.; Hoepken, B.; Arledge, T.; et al. Effect of Certolizumab Pegol on Signs and Symptoms in Patients with Psoriatic Arthritis: 24-Week Results of a Phase 3 Double-Blind Randomised Placebo-Controlled Study (RAPID-PsA). Ann. Rheum. Dis. 2014, 73, 48–55. [Google Scholar] [CrossRef]
- Lemos, L.L.P.; de Oliveira Costa, J.; Almeida, A.M.; Junior, H.O.; Barbosa, M.M.; Kakehasi, A.M.; Acurcio, F.A. Treatment of Psoriatic Arthritis with Anti-TNF Agents: A Systematic Review and Meta-Analysis of Efficacy, Effectiveness and Safety. Rheumatol. Int. 2014, 34, 1345–1360. [Google Scholar] [CrossRef]
- Rodgers, M.; Epstein, D.; Bojke, L.; Yang, H.; Craig, D.; Fonseca, T.; Myers, L.; Bruce, I.; Chalmers, R.; Bujkiewicz, S.; et al. Etanercept, Infliximab and Adalimumab for the Treatment of Psoriatic Arthritis: A Systematic Review and Economic Evaluation. Health Technol. Assess 2011, 15, 134. [Google Scholar] [CrossRef] [Green Version]
- Fénix-Caballero, S.; Alegre-Del Rey, E.J.; Castaño-Lara, R.; Puigventõs-Latorre, F.; Borrero-Rubio, J.M.; Lõpez-Vallejo, J.F. Direct and Indirect Comparison of the Efficacy and Safety of Adalimumab, Etanercept, Infliximab and Golimumab in Psoriatic Arthritis. J. Clin. Pharm. Ther. 2013, 38, 286–293. [Google Scholar] [CrossRef]
- Mease, P.J.; Gladman, D.D.; Collier, D.H.; Ritchlin, C.T.; Helliwell, P.S.; Liu, L.; Kricorian, G.; Chung, J.B. Etanercept and Methotrexate as Monotherapy or in Combination for Psoriatic Arthritis: Primary Results From a Randomized, Controlled Phase III Trial. Arthritis Rheumatol. 2019, 71, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Woolacott, N.; Vergel, Y.B.; Hawkins, N.; Kainth, A.; Khadjesari, Z.; Misso, K.; Light, K.; Asseburg, C.; Palmer, S.; Claxton, K.; et al. Etanercept and Infliximab for the Treatment of Psoriatic Arthritis: A Systematic Review and Economic Evaluation. Health Technol. Assess 2006, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendling, D.; Joshi, A.; Reilly, P.; Jalundhwala, Y.J.; Mittal, M.; Bao, Y. Comparing the Risk of Developing Uveitis in Patients Initiating Anti-Tumor Necrosis Factor Therapy for Ankylosing Spondylitis: An Analysis of a Large US Claims Database. Curr. Med. Res. Opin. 2014, 30, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Tamimoto, Y.; Kimoto, Y.; Uchino, A.; To, K.; Harashima, S.I.; Hatta, N.; Harada, M. Mechanisms for Cytotoxic Effects of Anti-Tumor Necrosis Factor Agents on Transmembrane Tumor Necrosis Factor Alpha-Expressing Cells: Comparison among Infliximab, Etanercept, and Adalimumab. Arthritis Rheum. 2008, 58, 1248–1257. [Google Scholar] [CrossRef]
- Minozzi, S.; Bonovas, S.; Lytras, T.; Pecoraro, V.; González-Lorenzo, M.; Bastiampillai, A.J.; Gabrielli, E.M.; Lonati, A.C.; Moja, L.; Cinquini, M.; et al. Risk of Infections Using Anti-TNF Agents in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis: A Systematic Review and Meta-Analysis. Expert Opin. Drug Saf. 2016, 15, 1243–1252. [Google Scholar] [CrossRef]
- Li, X.; Andersen, K.M.; Chang, H.Y.; Curtis, J.R.; Alexander, G.C. Comparative Risk of Serious Infections among Real-World Users of Biologics for Psoriasis or Psoriatic Arthritis. Ann. Rheum. Dis. 2020, 79, 285–291. [Google Scholar] [CrossRef]
- Seror, R.; Richez, C.; Sordet, C.; Rist, S.; Gossec, L.; Direz, G.; Houvenagel, E.; Berthelot, J.M.; Pagnoux, C.; Dernis, E.; et al. Pattern of Demyelination Occurring during Anti-TNF-α Therapy: A French National Survey. Rheumatology 2013, 52, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Leone, G.M.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; Fagone, P. Past, Present and (Foreseeable) Future of Biological Anti-TNF Alpha Therapy. J. Clin. Med. 2023, 12, 1630. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and Safety of Ustekinumab in Patients with Active Psoriatic Arthritis: 1 Year Results of the Phase 3, Multicentre, Double-Blind, Placebo-Controlled PSUMMIT 1 Trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; McInnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.K.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and Safety of the Anti-IL-12/23 P40 Monoclonal Antibody, Ustekinumab, in Patients with Active Psoriatic Arthritis despite Conventional Non-Biological and Biological Anti-Tumour Necrosis Factor Therapy: 6-Month and 1-Year Results of the Phase 3, Multicentre, Double-Blind, Placebo-Controlled, Randomised PSUMMIT 2 Trial. Ann. Rheum. Dis. 2014, 73, 990–999. [Google Scholar] [CrossRef]
- Azuaga, A.B.; Frade-Sosa, B.; Laiz, A.; Estrada, P.; Prior-Español, A.; Horcada, L.; Polino, L.; Moreno, M.; Moragues, C.; Urruticoechea-Arana, A.; et al. Effectiveness of Ustekinumab in Patients with Psoriatic Arthritis in a Real-World, Multicenter Study. Clin. Rheumatol. 2020, 39, 2963–2971. [Google Scholar] [CrossRef]
- Deodhar, A.; Helliwell, P.S.; Boehncke, W.H.; Kollmeier, A.P.; Hsia, E.C.; Subramanian, R.A.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; et al. Guselkumab in Patients with Active Psoriatic Arthritis Who Were Biologic-Naive or Had Previously Received TNFα Inhibitor Treatment (DISCOVER-1): A Double-Blind, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2020, 395, 1115–1125. [Google Scholar] [CrossRef]
- Mease, P.J.; Rahman, P.; Gottlieb, A.B.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; Zhuang, Y.; et al. Guselkumab in Biologic-Naive Patients with Active Psoriatic Arthritis (DISCOVER-2): A Double-Blind, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2020, 395, 1126–1136. [Google Scholar] [CrossRef]
- Coates, L.C.; Gossec, L.; Theander, E.; Bergmans, P.; Neuhold, M.; Karyekar, C.S.; Shawi, M.; Noël, W.; Schett, G.; McInnes, I.B. Efficacy and Safety of Guselkumab in Patients with Active Psoriatic Arthritis Who Are Inadequate Responders to Tumour Necrosis Factor Inhibitors: Results through One Year of a Phase IIIb, Randomised, Controlled Study (COSMOS). Ann. Rheum. Dis. 2022, 81, 359–369. [Google Scholar] [CrossRef]
- Kristensen, L.E.; Keiserman, M.; Papp, K.; McCasland, L.; White, D.; Lu, W.; Wang, Z.; Soliman, A.M.; Eldred, A.; Barcomb, L.; et al. Efficacy and Safety of Risankizumab for Active Psoriatic Arthritis: 24-Week Results from the Randomised, Double-Blind, Phase 3 KEEPsAKE 1 Trial. Ann. Rheum. Dis. 2022, 81, 225–231. [Google Scholar] [CrossRef]
- Östör, A.; van den Bosch, F.; Papp, K.; Asnal, C.; Blanco, R.; Aelion, J.; Alperovich, G.; Lu, W.; Wang, Z.; Soliman, A.M.; et al. Efficacy and Safety of Risankizumab for Active Psoriatic Arthritis: 24-Week Results from the Randomised, Double-Blind, Phase 3 KEEPsAKE 2 Trial. Ann. Rheum. Dis. 2022, 81, 351–358. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Identifier: NCT04314544. Available online: https://clinicaltrials.gov/ct2/show/NCT04314544 (accessed on 10 December 2022).
- ClinicalTrials.Gov. Identifier: NCT04314531. Available online: https://clinicaltrials.gov/ct2/show/NCT04314531 (accessed on 10 December 2022).
- Mease, P.J.; Chohan, S.; Fructuoso, F.J.G.; Luggen, M.E.; Rahman, P.; Raychaudhuri, S.P.; Chou, R.C.; Mendelsohn, A.M.; Rozzo, S.J.; Gottlieb, A. Efficacy and Safety of Tildrakizumab in Patients with Active Psoriatic Arthritis: Results of a Randomised, Double-Blind, Placebo-Controlled, Multiple-Dose, 52-Week Phase IIb Study. Ann. Rheum. Dis. 2021, 80, 1147–1157. [Google Scholar] [CrossRef]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Østergaard, M.; Wei, J.C.C.; Sieper, J.; Järvinen, P.; Tam, L.S.; Salvarani, C.; Kim, T.H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 Inhibitor, for Ankylosing Spondylitis: Results of a Randomised, Double-Blind, Placebo-Controlled, Proof-of-Concept, Dose-Finding Phase 2 Study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; van den Bosch, F.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an Interleukin-17A Antagonist in the Treatment of Ankylosing Spondylitis or Radiographic Axial Spondyloarthritis in Patients Previously Untreated with Biological Disease-Modifying Anti-Rheumatic Drugs (COAST-V): 16 Week Results of a Phase 3 Randomised, Double-Blind, Active-Controlled and Placebo-Controlled Trial. Lancet 2018, 392, 2441–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenna, T.J.; Davidson, S.I.; Duan, R.; Bradbury, L.A.; McFarlane, J.; Smith, M.; Weedon, H.; Street, S.; Thomas, R.; Thomas, G.P.; et al. Enrichment of Circulating Interleukin-17-Secreting Interleukin-23 Receptor-Positive γ/δ T Cells in Patients with Active Ankylosing Spondylitis. Arthritis Rheum. 2012, 64, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; la Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Helliwell, P.S.; Gladman, D.D.; Poddubnyy, D.; Baraliakos, X.; Chakravarty, S.D.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; et al. Efficacy of Guselkumab on Axial Involvement in Patients with Active Psoriatic Arthritis and Sacroiliitis: A Post-Hoc Analysis of the Phase 3 DISCOVER-1 and DISCOVER-2 Studies. Lancet Rheumatol. 2021, 3, e715–e723. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Identifier: NCT04929210. Available online: https://clinicaltrials.gov/ct2/show/NCT04929210 (accessed on 10 December 2022).
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; van der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J.; et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; van der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a Human Anti-Interleukin-17A Monoclonal Antibody, in Patients with Psoriatic Arthritis (FUTURE 2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Nash, P.; Mease, P.J.; McInnes, I.B.; Rahman, P.; Ritchlin, C.T.; Blanco, R.; Dokoupilova, E.; Andersson, M.; Kajekar, R.; Mpofu, S.; et al. Efficacy and Safety of Secukinumab Administration by Autoinjector in Patients with Psoriatic Arthritis: Results from a Randomized, Placebo-Controlled Trial (FUTURE 3). Arthritis Res. Ther. 2018, 20, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kivitz, A.J.; Nash, P.; Tahir, H.; Everding, A.; Mann, H.; Kaszuba, A.; Pellet, P.; Widmer, A.; Pricop, L.; Abrams, K. Efficacy and Safety of Subcutaneous Secukinumab 150 Mg with or Without Loading Regimen in Psoriatic Arthritis: Results from the FUTURE 4 Study. Rheumatol. Ther. 2019, 6, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; van der Heijde, D.; Landewé, R.; Mpofu, S.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Meiser, K.; et al. Secukinumab Improves Active Psoriatic Arthritis Symptoms and Inhibits Radiographic Progression: Primary Results from the Randomised, Double-Blind, Phase III FUTURE 5 Study. Ann. Rheum. Dis. 2018, 77, 890–897. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Behrens, F.; Mease, P.J.; Kavanaugh, A.; Ritchlin, C.; Nash, P.; Masmitja, J.G.; Goupille, P.; Korotaeva, T.; Gottlieb, A.B.; et al. Secukinumab versus Adalimumab for Treatment of Active Psoriatic Arthritis (EXCEED): A Double-Blind, Parallel-Group, Randomised, Active-Controlled, Phase 3b Trial. Lancet 2020, 395, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Deodhar, A.; McInnes, I.B.; Baraliakos, X.; Reich, K.; Schreiber, S.; Bao, W.; Marfo, K.; Richards, H.B.; Pricop, L.; et al. Long-Term Safety of Secukinumab Over Five Years in Patients with Moderate-to-Severe Plaque Psoriasis, Psoriatic Arthritis and Ankylosing Spondylitis: Update on Integrated Pooled Clinical Trial and Post-Marketing Surveillance Data. Acta Derm. Venereol. 2022, 102, adv00698. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D. Ixekizumab, an Interleukin-17A Specific Monoclonal Antibody, for the Treatment of Biologic-Naive Patients with Active Psoriatic Arthritis: Results from the 24-Week Randomised, Double-Blind, Placebo-Controlled and Active (Adalimumab)-Controlled Period of the Phase III Trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, P.; Kirkham, B.; Okada, M.; Rahman, P.; Combe, B.; Burmester, G.R.; Adams, D.H.; Kerr, L.; Lee, C.; Shuler, C.L.; et al. Ixekizumab for the Treatment of Patients with Active Psoriatic Arthritis and an Inadequate Response to Tumour Necrosis Factor Inhibitors: Results from the 24-Week Randomised, Double-Blind, Placebo-Controlled Period of the SPIRIT-P2 Phase 3 Trial. Lancet 2017, 389, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Smolen, J.S.; Behrens, F.; Nash, P.; Liu Leage, S.; Li, L.; Tahir, H.; Gooderham, M.; Krishnan, E.; Liu-Seifert, H.; et al. A Head-to-Head Comparison of the Efficacy and Safety of Ixekizumab and Adalimumab in Biological-Naïve Patients with Active Psoriatic Arthritis: 24-Week Results of a Randomised, Open-Label, Blinded-Assessor Trial. Ann. Rheum. Dis. 2020, 79, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGonagle, D.G.; McInnes, I.B.; Kirkham, B.W.; Sherlock, J.; Moots, R. The Role of IL-17A in Axial Spondyloarthritis and Psoriatic Arthritis: Recent Advances and Controversies. Ann. Rheum. Dis. 2019, 78, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Merola, J.F.; Landewé, R.; McInnes, I.B.; Mease, P.J.; Ritchlin, C.T.; Tanaka, Y.; Asahina, A.; Behrens, F.; Gladman, D.D.; Gossec, L.; et al. Bimekizumab in Patients with Active Psoriatic Arthritis and Previous Inadequate Response or Intolerance to Tumour Necrosis Factor-α Inhibitors: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial (BE COMPLETE). Lancet 2023, 401, 38–48. [Google Scholar] [CrossRef]
- McInnes, I.B.; Asahina, A.; Coates, L.C.; Landewé, R.; Merola, J.F.; Ritchlin, C.T.; Tanaka, Y.; Gossec, L.; Gottlieb, A.B.; Warren, R.B.; et al. Bimekizumab in Patients with Psoriatic Arthritis, Naive to Biologic Treatment: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial (BE OPTIMAL). Lancet 2023, 401, 25–37. [Google Scholar] [CrossRef]
- Lebwohl, M.; Leonardi, C.; Armstrong, A.; Rawnsley, N.; Alexander, B.; Goehring, E.; Kerdel, F.; Jacobson, A. Three-Year U.S. Pharmacovigilance Report of Brodalumab. Dermatol. Ther. 2021, 34, e15105. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a Human Anti-IL-17A Monoclonal Antibody, for Moderate to Severe Crohn’s Disease: Unexpected Results of a Randomised, Double-Blind Placebo-Controlled Trial. Gut 2012, 61, 1693. [Google Scholar] [CrossRef] [Green Version]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients with Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.F.; Sendid, B.; Jouault, T.; Poulain, D. Secukinumab Failure in Crohn’s Disease: The Yeast Connection? Gut 2013, 62, 800–801. [Google Scholar] [CrossRef] [PubMed]
- Traves, P.G.; Murray, B.; Campigotto, F.; Galien, R.; Meng, A.; di Paolo, J.A. JAK Selectivity and the Implications for Clinical Inhibition of Pharmacodynamic Cytokine Signalling by Filgotinib, Upadacitinib, Tofacitinib and Baricitinib. Ann. Rheum. Dis. 2021, 80, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.; Rigby, W.; Azevedo, V.F.; Behrens, F.; Blanco, R.; Kaszuba, A.; Kudlacz, E.; Wang, C.; Menon, S.; Hendrikx, T.; et al. Tofacitinib for Psoriatic Arthritis in Patients with an Inadequate Response to TNF Inhibitors. N. Engl. J. Med. 2017, 377, 1525–1536. [Google Scholar] [CrossRef]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Kato, K.; Magrey, M.; Merola, J.F.; Kishimoto, M.; Pacheco-Tena, C.; Haaland, D.; Chen, L.; Duan, Y.; Zueger, P.; et al. Upadacitinib in Patients with Psoriatic Arthritis and an Inadequate Response to Non-Biological Therapy: 56-Week Data from the Phase 3 SELECT-PsA 1 Study. RMD Open 2021, 7, e001838. [Google Scholar] [CrossRef]
- Mease, P.J.; Lertratanakul, A.; Anderson, J.K.; Papp, K.; van den Bosch, F.; Tsuji, S.; Dokoupilova, E.; Keiserman, M.; Wang, X.; Zhong, S.; et al. Upadacitinib for Psoriatic Arthritis Refractory to Biologics: SELECT-PsA 2. Ann. Rheum. Dis. 2021, 80, 312–320. [Google Scholar] [CrossRef]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and Safety of Filgotinib, a Selective Janus Kinase 1 Inhibitor, in Patients with Active Psoriatic Arthritis (EQUATOR): Results from a Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Identifier: NCT04115748. Available online: https://clinicaltrials.gov/ct2/show/NCT04115748 (accessed on 10 December 2022).
- ClinicalTrials.Gov. Identifier: NCT04115839. Available online: https://clinicaltrials.gov/ct2/show/NCT04115839 (accessed on 10 December 2022).
- Mease, P.J.; Deodhar, A.A.; van der Heijde, D.; Behrens, F.; Kivitz, A.J.; Neal, J.; Kim, J.; Singhal, S.; Nowak, M.; Banerjee, S. Efficacy and Safety of Selective TYK2 Inhibitor, Deucravacitinib, in a Phase II Trial in Psoriatic Arthritis. Ann. Rheum. Dis. 2022, 81, 815–822. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Identifier: NCT04908202. Available online: https://clinicaltrials.gov/ct2/show/NCT04908202 (accessed on 10 December 2022).
- ClinicalTrials.Gov. Identifier: NCT04908189. Available online: https://clinicaltrials.gov/ct2/show/NCT04908189 (accessed on 10 December 2022).
- Mease, P.; Helliwell, P.S.; Silwinska-Stanczyk, P.; Miakisz, M.; Ostor, A.; Peeva, E.; Vincent, M.; Sikirica, V.; Winnette, R.; Qiu, R. Efficacy and Safety of Brepocitinib (Tyrosine Kinase 2/Janus Kinase 1 Inhibitor) for the Treatment of Active Psoriatic Arthritis: Results from a Phase 2b Randomized Controlled Trial. Arthritis Rheumatol. 2021, 73, 1009–1011. [Google Scholar]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus Tocilizumab in Anti-TNF Inadequate Responder Patients with Rheumatoid Arthritis (R4RA): 16-Week Outcomes of a Stratified, Biopsy-Driven, Multicentre, Open-Label, Phase 4 Randomised Controlled Trial. Lancet 2021, 397, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Surace, A.E.A.; Goldmann, K.; Sciacca, E.; Çubuk, C.; Giorli, G.; John, C.R.; Nerviani, A.; Fossati-Jimack, L.; Thorborn, G.; et al. Rituximab versus Tocilizumab in Rheumatoid Arthritis: Synovial Biopsy-Based Biomarker Analysis of the Phase 4 R4RA Randomized Trial. Nat. Med. 2022, 28, 1256–1268. [Google Scholar] [CrossRef]
- Gurke, R.; Bendes, A.; Bowes, J.; Koehm, M.; Twyman, R.M.; Barton, A.; Elewaut, D.; Goodyear, C.; Hahnefeld, L.; Hillenbrand, R.; et al. Omics and Multi-Omics Analysis for the Early Identification and Improved Outcome of Patients with Psoriatic Arthritis. Biomedicines 2022, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, O.; Pennington, S.R. HIPPOCRATES: Improving Diagnosis and Outcomes in Psoriatic Arthritis. Nat. Rev. Rheumatol. 2022, 18, 123–124. [Google Scholar] [CrossRef]
- Winthrop, K.L.; Isaacs, J.D.; Mease, P.J.; Boumpas, D.T.; Baraliakos, X.; Gottenberg, J.-E.; Siebert, S.; Mosca, M.; Basu, N.; Orange, D.; et al. Unmet Need in Rheumatology: Reports from the Advances in Targeted Therapies Meeting, 2022. Ann. Rheum. Dis. 2023. online first. [Google Scholar] [CrossRef]
- Kuwert, T.; Schmidkonz, C.; Prante, O.; Schett, G.; Ramming, A. FAPI PET Opens a New Window to Understanding Immune-Mediated Inflammatory Diseases. J. Nucl. Med. 2022, 63, 1136–1137. [Google Scholar] [CrossRef]
- Haberman, R.H.; MacFarlane, K.A.; Catron, S.; Samuels, J.; Blank, R.B.; Toprover, M.; Uddin, Z.; Hu, J.; Castillo, R.; Gong, C.; et al. Efficacy of Guselkumab, a Selective IL-23 Inhibitor, in Preventing Arthritis in a Multicentre Psoriasis At-Risk Cohort (PAMPA): Protocol of a Randomised, Double-Blind, Placebo Controlled Multicentre Trial. BMJ Open 2022, 12, e063650. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azuaga, A.B.; Ramírez, J.; Cañete, J.D. Psoriatic Arthritis: Pathogenesis and Targeted Therapies. Int. J. Mol. Sci. 2023, 24, 4901. https://doi.org/10.3390/ijms24054901
Azuaga AB, Ramírez J, Cañete JD. Psoriatic Arthritis: Pathogenesis and Targeted Therapies. International Journal of Molecular Sciences. 2023; 24(5):4901. https://doi.org/10.3390/ijms24054901
Chicago/Turabian StyleAzuaga, Ana Belén, Julio Ramírez, and Juan D. Cañete. 2023. "Psoriatic Arthritis: Pathogenesis and Targeted Therapies" International Journal of Molecular Sciences 24, no. 5: 4901. https://doi.org/10.3390/ijms24054901