

Simple In Vitro 18O Labeling for Improved Mass Spectrometry-Based Drug Metabolites Identification: Deep Drug Metabolism Study
 , , and
, , and
Abstract
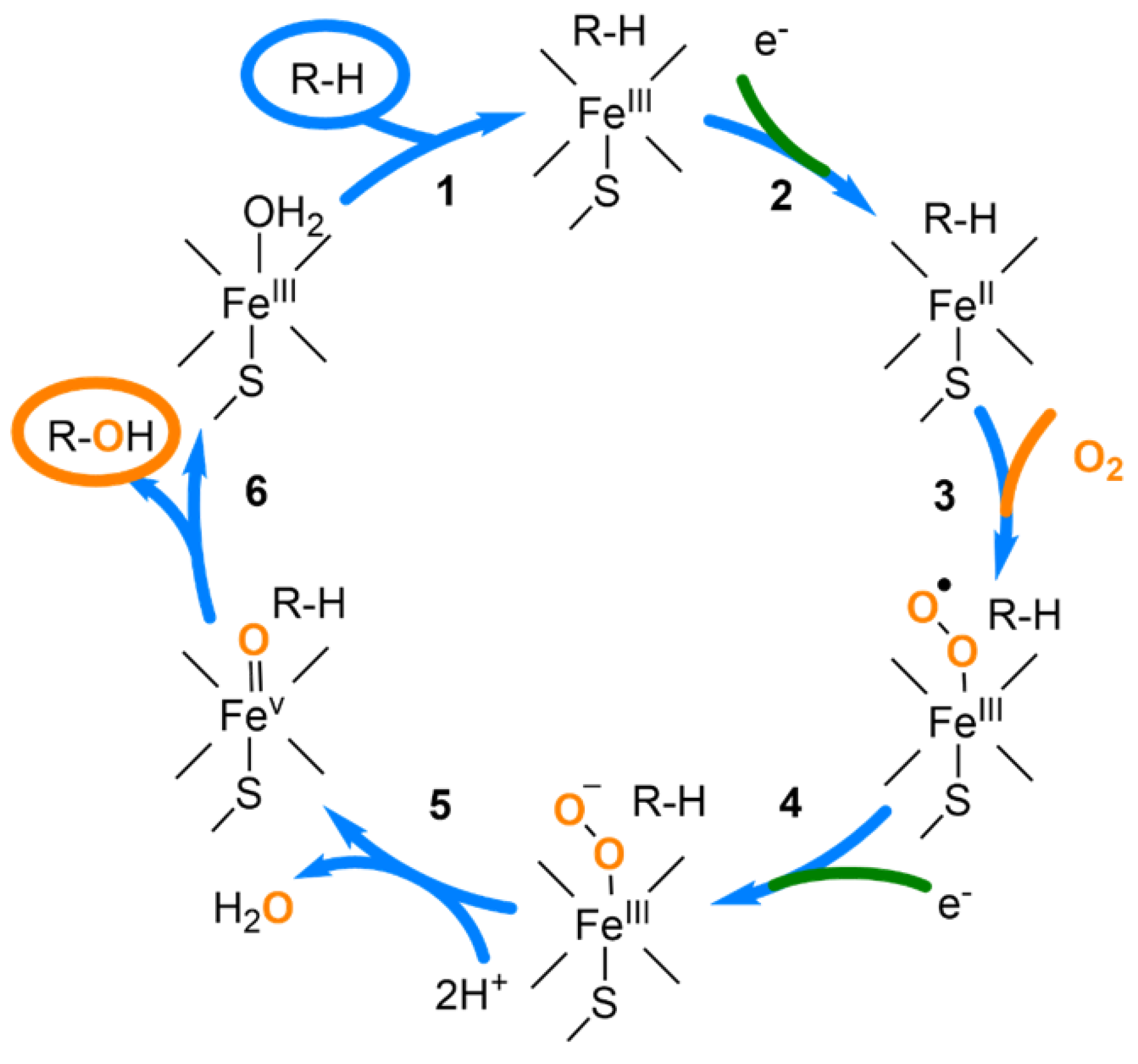
:1. Introduction
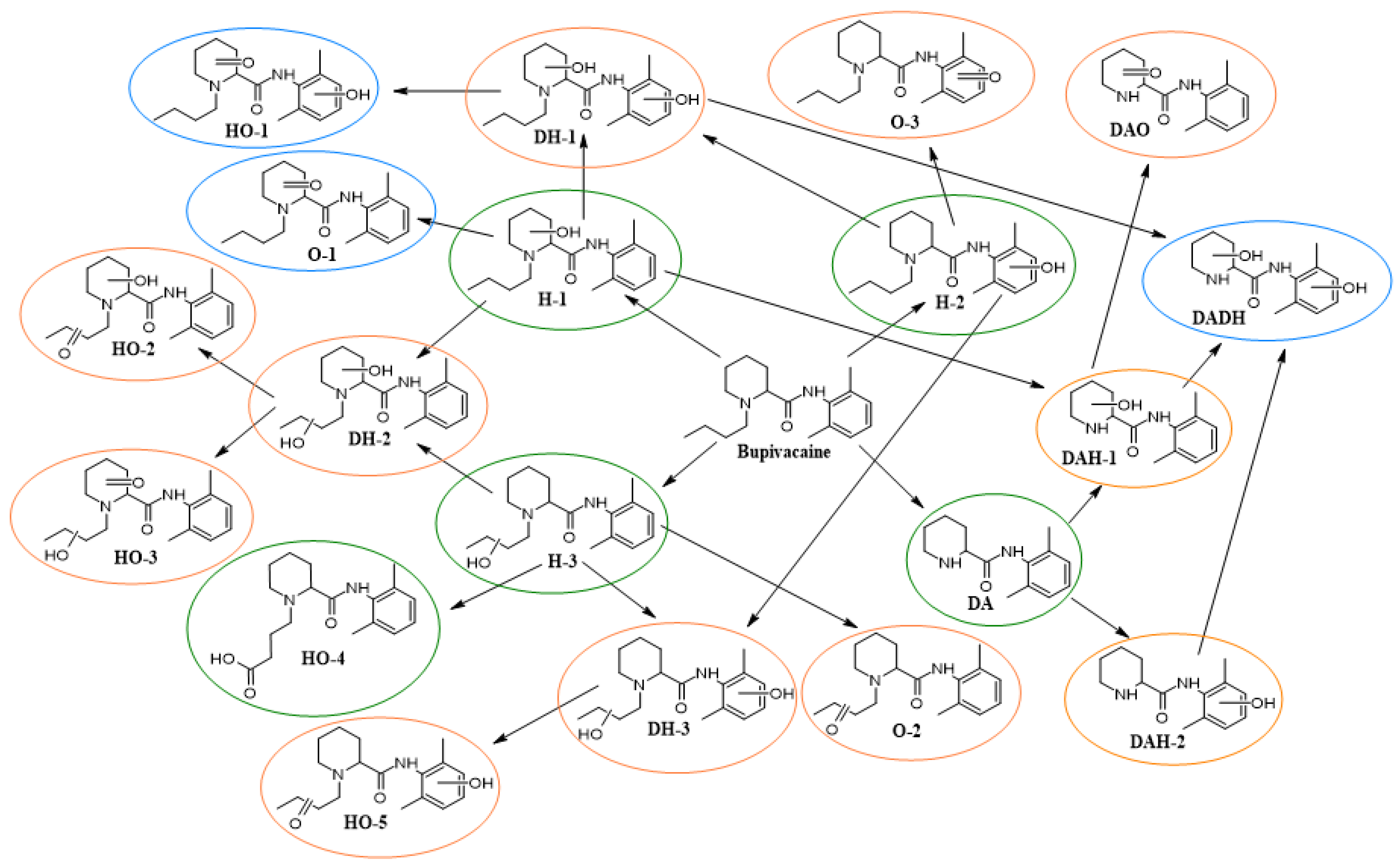
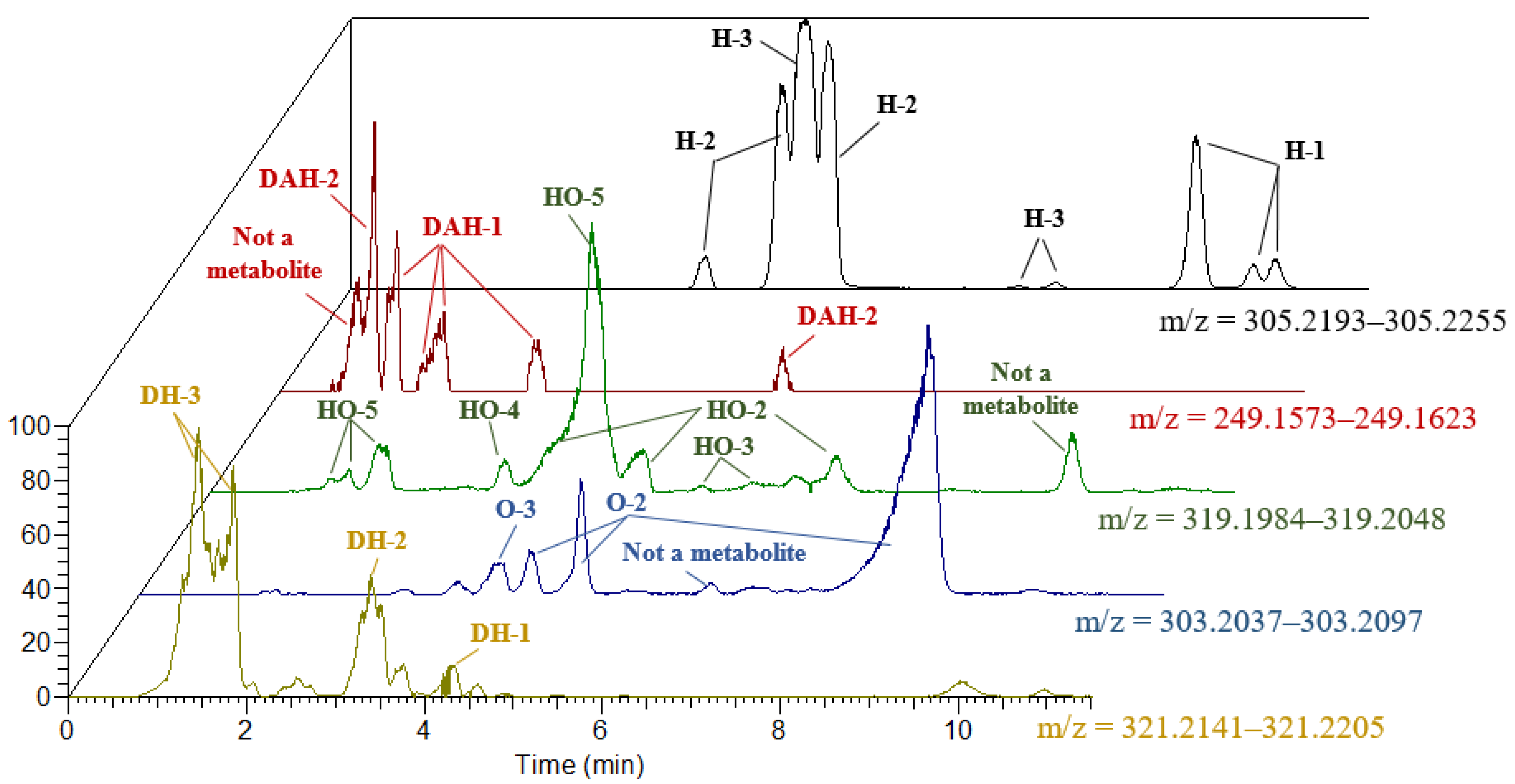
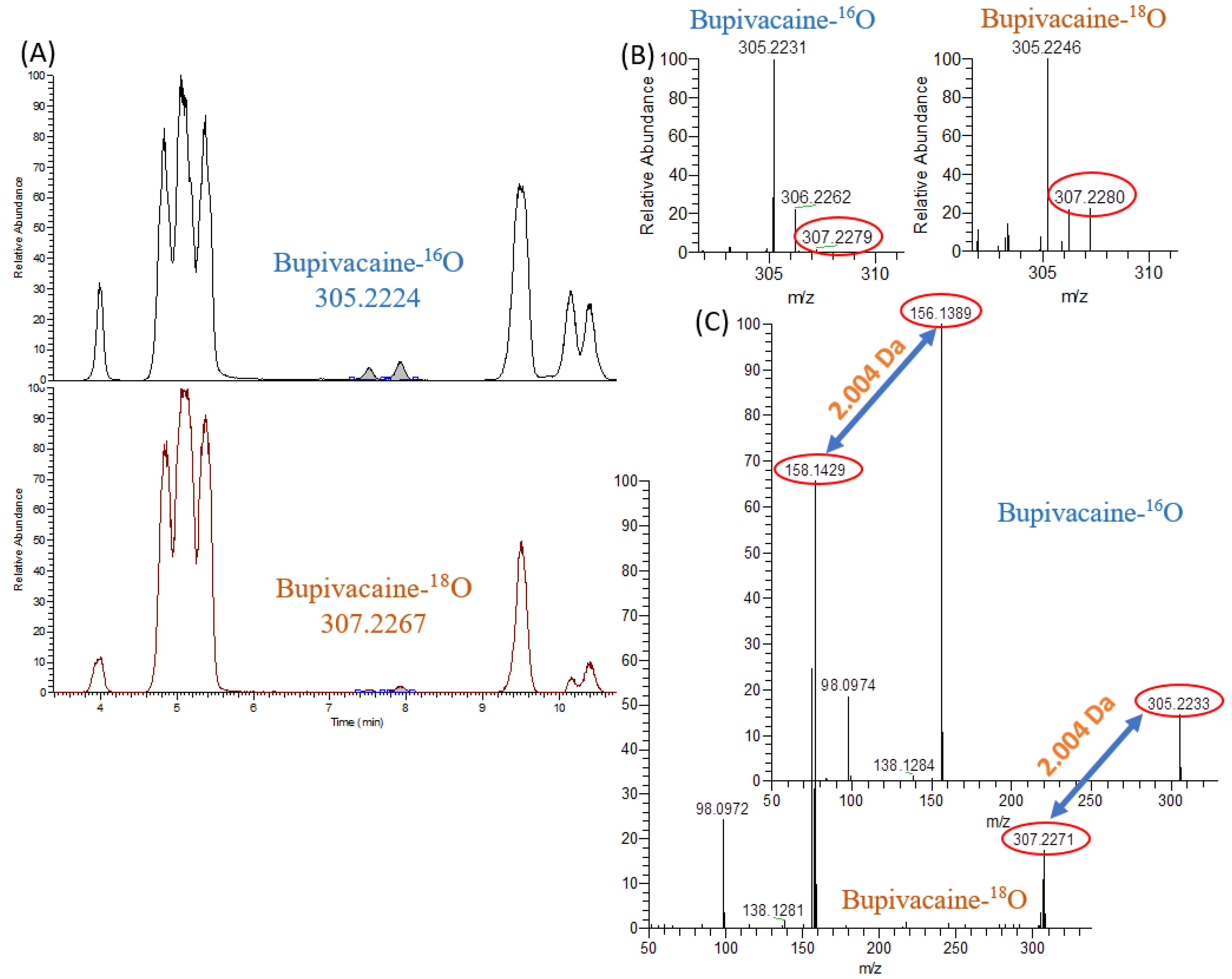
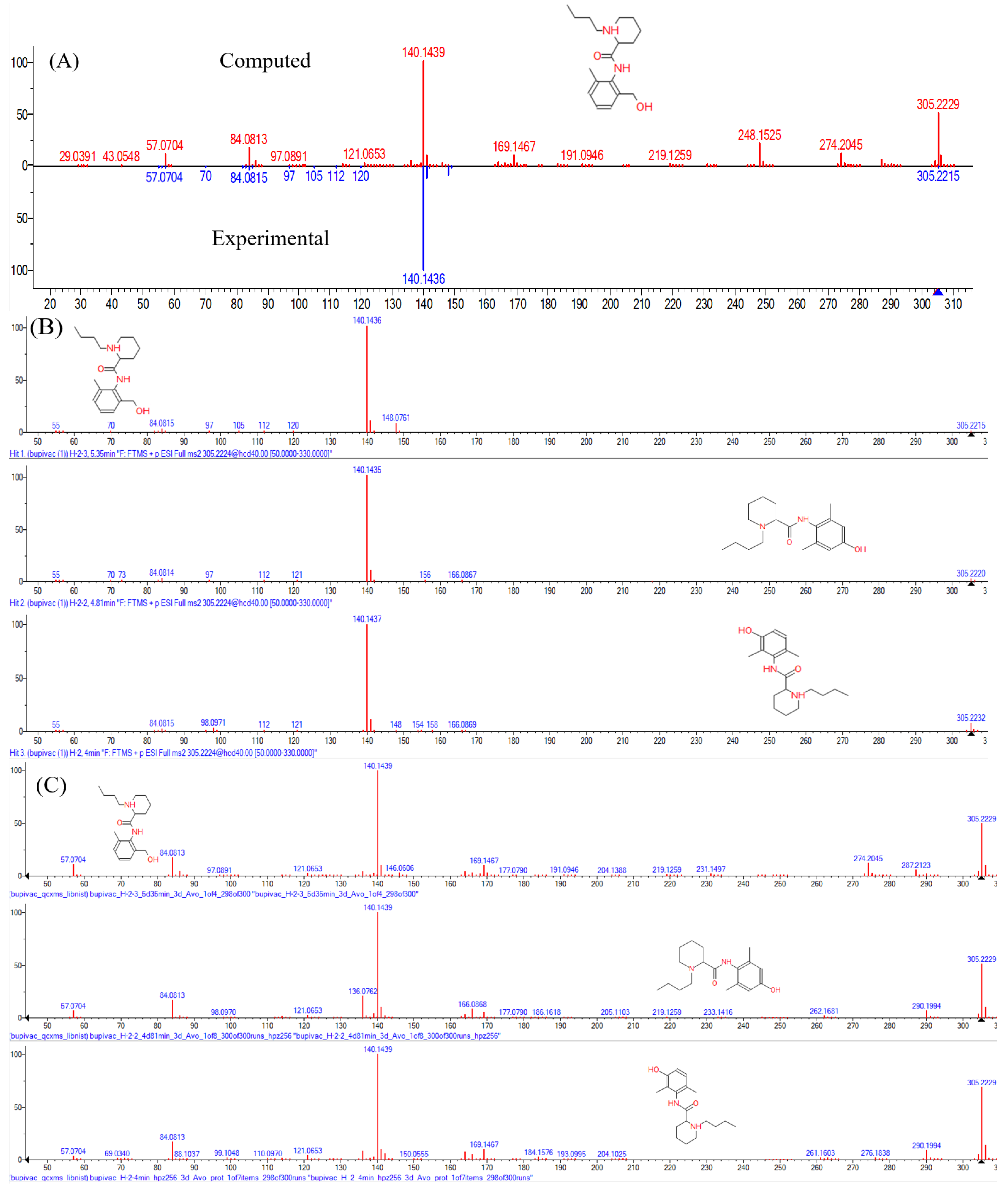
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Reagents
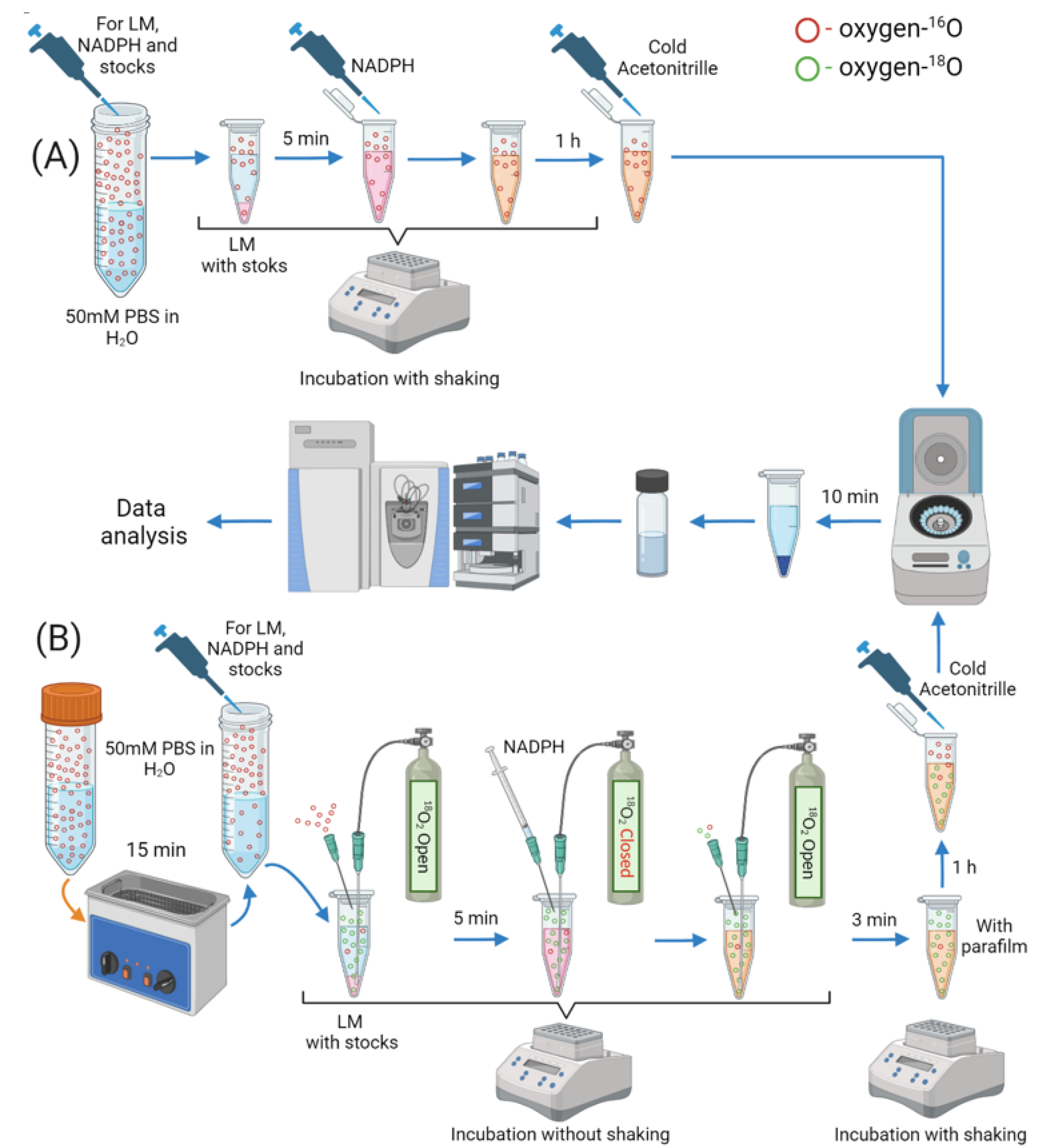
3.2. Experiment
3.3. Sample Preparation
3.4. HPLC-HRMS Conditions
3.5. Oxygen-18 Label Stability Study
3.6. Data Analysis
4. Conclusions
Associated Content
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Tang, W. Drug Metabolism in Drug Discovery and Development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Research and Development in the Pharmaceutical Industry|Congressional Budget Office. Available online: https://www.cbo.gov/publication/57126 (accessed on 13 December 2022).
- Zhang, Z.; Zhu, M.; Tang, W. Metabolite Identification and Profiling in Drug Design: Current Practice and Future Directions. Curr. Pharm. Des. 2009, 15, 2220–2235. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical Experimental Models of Drug Metabolism and Disposition in Drug Discovery and Development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7.8.1–7.8.24. [Google Scholar] [CrossRef]
- Esteves, F.; Rueff, J.; Kranendonk, M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. J. Xenobiotics 2021, 11, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, N.; Tian, X.; Liu, T.; Fang, Y.; Zhou, J.; Wen, Q.; Xu, B.; Qi, B.; Gao, J.; et al. Content and Activity of Human Liver Microsomal Protein and Prediction of Individual Hepatic Clearance in Vivo. Sci. Rep. 2015, 5, 17671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfmacher, W. High-Resolution Mass Spectrometry Will Dramatically Change Our Drug-Discovery Bioanalysis Procedures. Bioanalysis 2011, 3, 1169–1171. [Google Scholar] [CrossRef]
- Lu, J.-D.; Xue, J. Poisoning: Kinetics to Therapeutics. In Critical Care Nephrology, 3rd ed; Elsevier: Amsterdam, The Netherlands, 2017; pp. 600–629.e7. [Google Scholar]
- Hao, L.; Wang, J.; Page, D.; Asthana, S.; Zetterberg, H.; Carlsson, C.; Okonkwo, O.C.; Li, L. Comparative Evaluation of MS-Based Metabolomics Software and Its Application to Preclinical Alzheimer’s Disease. Sci. Rep. 2018, 8, 9291. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A Database-Searching Algorithm for Metabolite Identification in Metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag Relaunched: Incorporating Strategies beyond in Silico Fragmentation. J. Cheminformatics 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PyFragMS─A Web Tool for the Investigation of the Collision-Induced Fragmentation Pathways|ACS Omega. Available online: https://pubs.acs.org/doi/full/10.1021/acsomega.1c07272 (accessed on 29 January 2023).
- Kostyukevich, Y.; Zherebker, A.; Orlov, A.; Kovaleva, O.; Burykina, T.; Isotov, B.; Nikolaev, E.N. Hydrogen/Deuterium and 16O/18O-Exchange Mass Spectrometry Boosting the Reliability of Compound Identification. Anal. Chem. 2020, 92, 6877–6885. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Zehl, M.; Jørgensen, T.J.D. Measuring the Hydrogen/Deuterium Exchange of Proteins at High Spatial Resolution by Mass Spectrometry: Overcoming Gas-Phase Hydrogen/Deuterium Scrambling. Acc. Chem. Res. 2014, 47, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.K.; Broadhurst, R.W.; Skelton, P.C.; Arkin, I.T. Hydrogen/Deuterium Exchange of Hydrophobic Peptides in Model Membranes by Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2002, 13, 1376–1387. [Google Scholar] [CrossRef] [Green Version]
- Damont, A.; Legrand, A.; Cao, C.; Fenaille, F.; Tabet, J.-C. Hydrogen/Deuterium Exchange Mass Spectrometry in the World of Small Molecules. Mass Spectrom. Rev. 2021, e21765. [Google Scholar] [CrossRef]
- Liu, D.Q.; Hop, C.E.C.A.; Beconi, M.G.; Mao, A.; Chiu, S.-H.L. Use of On-Line Hydrogen/Deuterium Exchange to Facilitate Metabolite Identification. Rapid Commun. Mass Spectrom. 2001, 15, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Padilha, E.C.; Wang, J.; Kerns, E.; Lee, A.; Huang, W.; Jiang, J.; McKew, J.; Mutlib, A.; Peccinini, R.G.; Yu, P.B.; et al. Application of in Vitro Drug Metabolism Studies in Chemical Structure Optimization for the Treatment of Fibrodysplasia Ossificans Progressiva (FOP). Front. Pharmacol. 2019, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Lam, W.; Ramanathan, R. In Electrospray Ionization Source Hydrogen/Deuterium Exchange LC-MS and LC-MS/MS for Characterization of Metabolites. J. Am. Soc. Mass Spectrom. 2002, 13, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.; Ramanathan, R. System and Method for Characterizing Metabolites Using Hydrogen/Deuterium Exchange 2003. U.S. Patent Application 10/387,613, 18 September 2003. [Google Scholar]
- Ye, X.; Luke, B.; Andresson, T.; Blonder, J. 18O Stable Isotope Labeling in MS-Based Proteomics. Brief. Funct. Genomic. Proteomic. 2009, 8, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Rumiantseva, L.; Osipenko, S.; Zharikov, A.; Kireev, A.; Nikolaev, E.N.; Kostyukevich, Y. Analysis of 16O/18O and H/D Exchange Reactions between Carbohydrates and Heavy Water Using High-Resolution Mass Spectrometry. Int. J. Mol. Sci. 2022, 23, 3585. [Google Scholar] [CrossRef]
- Osipenko, S.; Zherebker, A.; Rumiantseva, L.; Kovaleva, O.; Nikolaev, E.N.; Kostyukevich, Y. Oxygen Isotope Exchange Reaction for Untargeted LC–MS Analysis. J. Am. Soc. Mass Spectrom. 2022, 33, 390–398. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, Y.-T.; Xu, Z.-B.; Qu, J. Hot Water-Promoted Ring-Opening of Epoxides and Aziridines by Water and Other Nucleopliles. J. Org. Chem. 2008, 73, 2270–2274. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water". Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-J.; Chen, L. Organic Chemistry in Water. Chem Soc Rev 2006, 35, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.R.; Klein, P.D. A Selected Bibliography of Biomedical and Environmental Applications of Stable Isotopes. IV—17O,18O And34S 1971-1976. Biol. Mass Spectrom. 1978, 5, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Renson, J.; Weissbach, H.; Udenfriend, S. On the Mechanism of Oxidative Cleavage of Aryl-Alkyl Ethers by Liver Microsomes. Mol. Pharmacol. 1965, 1, 145–148. [Google Scholar] [PubMed]
- Murphy, P.J. Enzymatic Oxidation of Nicotine to Nicotine Δ1′(5′) Iminium Ion: A Newly Discovered Intermediate in the Metabolism of Nicotine. J. Biol. Chem. 1973, 248, 2796–2800. [Google Scholar] [CrossRef]
- John Parli, C.; Wang, N.; McMahon, R.E. The Mechanism of the Oxidation of D-Amphetamine by Rabbit Liver Oxygenase. Oxygen-18 Studies. Biochem. Biophys. Res. Commun. 1971, 43, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, W. In Vitro Microsomal Metabolic Studies on a Selective MGluR5 Antagonist MTEP: Characterization of In Vitro Metabolites and Identification of a Novel Thiazole Ring Opening Aldehyde Metabolite. Xenobiotica 2005, 35, 797–809. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Cytochrome P450 Substrate Oxidation: MiniReview. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef]
- Available online: Https://Www.Ema.Europa.Eu/En/Documents/Mrl-Report/Bupivacaine-Porcine-Species-Summary-Report-Committee-Veterinary-Medicinal-Products_en.Pdf (accessed on 1 February 2023).
- Rydevik, A.; Bondesson, U.; Hedeland, M. Structural Elucidation of Phase I and II Metabolites of Bupivacaine in Horse Urine and Fungi of the Cunninghamella Species Using Liquid Chromatography/Multi-Stage Mass Spectrometry: MS Study of the Metabolism of Bupivacaine in Horse and Fungus. Rapid Commun. Mass Spectrom. 2012, 26, 1338–1346. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN Small Molecule Dataset for Machine Learning-Based Retention Time Prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, E.S.; Matyushin, D.D.; Plyushchenko, I.V.; Stavrianidi, A.N.; Buryak, A.K. Deep Learning for Retention Time Prediction in Reversed-Phase Liquid Chromatography. J. Chromatogr. A 2022, 1664, 462792. [Google Scholar] [CrossRef]
- Ju, R.; Liu, X.; Zheng, F.; Lu, X.; Xu, G.; Lin, X. Deep Neural Network Pretrained by Weighted Autoencoders and Transfer Learning for Retention Time Prediction of Small Molecules. Anal. Chem. 2021, 93, 15651–15658. [Google Scholar] [CrossRef] [PubMed]
- Zaretckii, M.; Bashkirova, I.; Osipenko, S.; Kostyukevich, Y.; Nikolaev, E.; Popov, P. 3D Chemical Structures Allow Robust Deep Learning Models for Retention Time Prediction. Digit. Discov. 2022, 1, 711–718. [Google Scholar] [CrossRef]
- Osipenko, S.; Nikolaev, E.; Kostyukevich, Y. Retention Time Prediction with Message-Passing Neural Networks. Separations 2022, 9, 291. [Google Scholar] [CrossRef]
- Osipenko, S.; Botashev, K.; Nikolaev, E.; Kostyukevich, Y. Transfer Learning for Small Molecule Retention Predictions. J. Chromatogr. A 2021, 1644, 462119. [Google Scholar] [CrossRef] [PubMed]
- Osipenko, S.; Bashkirova, I.; Sosnin, S.; Kovaleva, O.; Fedorov, M.; Nikolaev, E.; Kostyukevich, Y. Machine Learning to Predict Retention Time of Small Molecules in Nano-HPLC. Anal. Bioanal. Chem. 2020, 412, 7767–7776. [Google Scholar] [CrossRef]
- Legonkova, L.F.; Bushma, M.I.; Zverinsky, I.V.; Eismont, K.A. Inhibition of Enzymes of Drug Metabolism in Rat Liver by Ultrasound and Correction by Heparin. Exp. Toxicol. Pathol. 1999, 51, 360–362. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef] [PubMed]
- Quantum Chemistry-Based Molecular Dynamics Simulations as a Tool for the Assignment of ESI-MS/MS Spectra of Drug Molecules—Schnegotzki—2022—Chemistry—A European Journal—Wiley Online Library. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/full/10.1002/chem.202200318 (accessed on 29 January 2023).
- Koopman, J.; Grimme, S. From QCEIMS to QCxMS: A Tool to Routinely Calculate CID Mass Spectra Using Molecular Dynamics. J. Am. Soc. Mass Spectrom. 2021, 32, 1735–1751. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes of Metabolites | RT of Detected Metabolites, min | [M+H]+, m/z | Characteristic Product Ions |
|---|---|---|---|
| H-1 | 9.50; 10.15; 10.40 | 305.2224 | 82.0659 (C5H8N); 96.0816 (C6H10N); 156.1393 (C9H18NO) |
| H-2 | 3.98; 4.82; 5.37 | 84.0815 (C5H10N); 98.0974 (C6H12N); 140.1440 (C9H18N) | |
| H-3 | 5.08; 7.51; 7.92 | 84.0815 (C5H10N); 98.0974 (C6H12N); 156.1393 (C9H18NO) | |
| DAH-1 | 1.30; 1.68; 1.83; 2.93 | 249.1598 | 82.0659 (C5H8N); 100.0764 (C5H10NO) |
| DAH-2 | 1.04; 6.38 | 84.0815 (C5H10N) | |
| HO-1 | not found | 319.2016 | not found |
| HO-2 | 4.25; 4.66; 7.00 | 82.0658 (C5H8N); 100.0764 (C5H10NO); 112.0763 (C6H10NO); 170.1180 (C;9H16NO2); 261.1604 (C15H21N2O2) | |
| HO-3 | 5.10; 5.47 | 82.0658 (C5H8N); 170.1180 (C;9H16NO2); 259.1444 (C15H19N2O2) | |
| HO-4 | 3.30 | 84.0815 (C5H10N); 96.0816 (C6H10N); 170.1180 (C;9H16NO2); 245.1653 (C15H21N2O) | |
| HO-5 | 1.25; 1.40; 2.00; 4.35 | 84.0815 (C5H10N); 96.0816 (C6H10N); 154.1231 (C9H16NO); 261.1603 (C15H21N2O2) | |
| O-1 | not found | 303.2067 | not found |
| O-2 | 3.30; 5.00; 8.83 | 84.0815 (C5H10N); 96.0816 (C6H10N); 154.1232 (C9H16NO); 245.1653 (C15H21N2O) | |
| O-3 | 3.00 | 84.0815 (C5H10N); 140.1440 (C9H18N) 259.1444 (C15H19N2O2) | |
| DH-1 | 4.10 | 321.2173 | 96.0816 (C6H10N); 156.1393 (C9H18NO) |
| DH-2 | 3.29 | 84.0815 (C5H10N); 114.0920 (C6H12NO); 172.1337 (C9H18NO2) | |
| DH-3 | 1.45; 1.87 | 84.0815 (C5H10N); 98.0974 (C6H12N); 156.1393 (C9H18NO) | |
| DAO | 5.61 | 247.1448 | 70.0658 (C4H8N); 98.0608 (C5H8NO); 122.0964 (C8H12N) |
| DADH | not found | not found | not found |
| DA | 3.06 | 233.1648 | 84.0815 (C5H10N); 150.0920 (C9H12NO) detected without isotope labeling |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tupertsev, B.; Osipenko, S.; Kireev, A.; Nikolaev, E.; Kostyukevich, Y. Simple In Vitro 18O Labeling for Improved Mass Spectrometry-Based Drug Metabolites Identification: Deep Drug Metabolism Study. Int. J. Mol. Sci. 2023, 24, 4569. https://doi.org/10.3390/ijms24054569
Tupertsev B, Osipenko S, Kireev A, Nikolaev E, Kostyukevich Y. Simple In Vitro 18O Labeling for Improved Mass Spectrometry-Based Drug Metabolites Identification: Deep Drug Metabolism Study. International Journal of Molecular Sciences. 2023; 24(5):4569. https://doi.org/10.3390/ijms24054569
Chicago/Turabian StyleTupertsev, Boris, Sergey Osipenko, Albert Kireev, Eugene Nikolaev, and Yury Kostyukevich. 2023. "Simple In Vitro 18O Labeling for Improved Mass Spectrometry-Based Drug Metabolites Identification: Deep Drug Metabolism Study" International Journal of Molecular Sciences 24, no. 5: 4569. https://doi.org/10.3390/ijms24054569