
Myostatin Overexpression and Smad Pathway in Detrusor Derived from Pediatric Patients with End-Stage Lower Urinary Tract Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
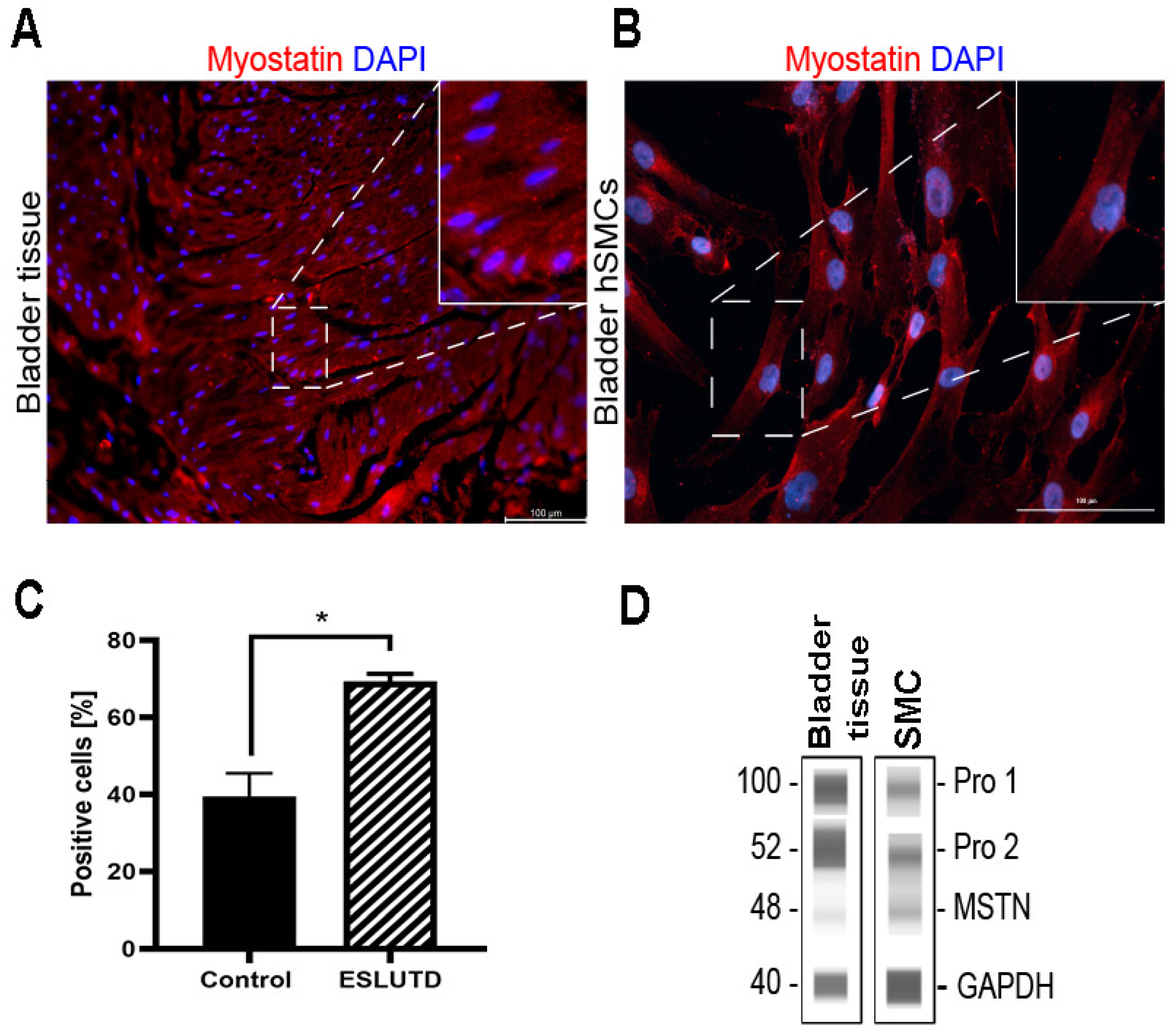
2.1. Myostatin Expression in Human Bladder
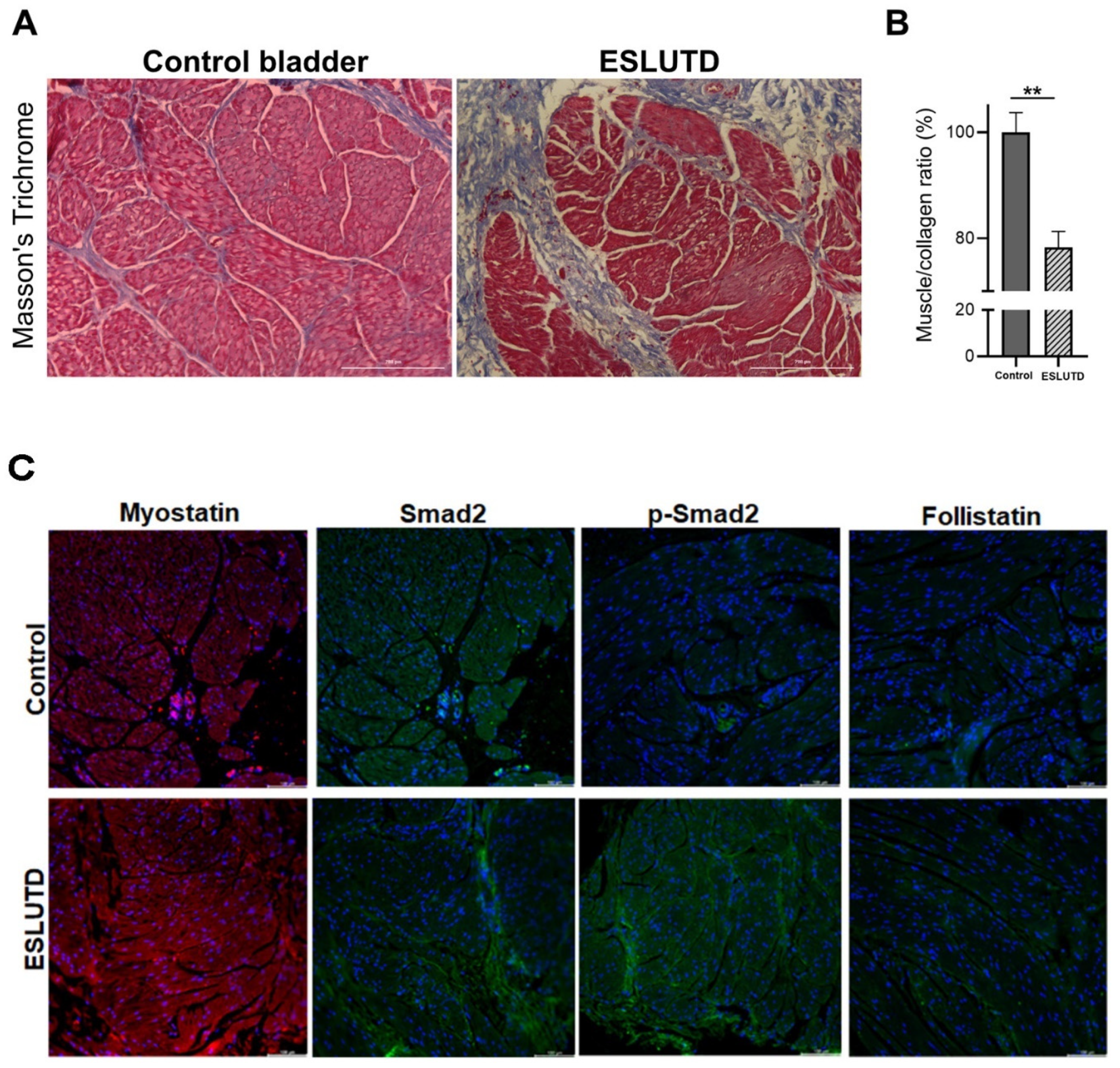
2.2. Comparison of Myostatin Expression in Human Bladder Tissue Derived from Control and ESLUTD Pediatric Patients
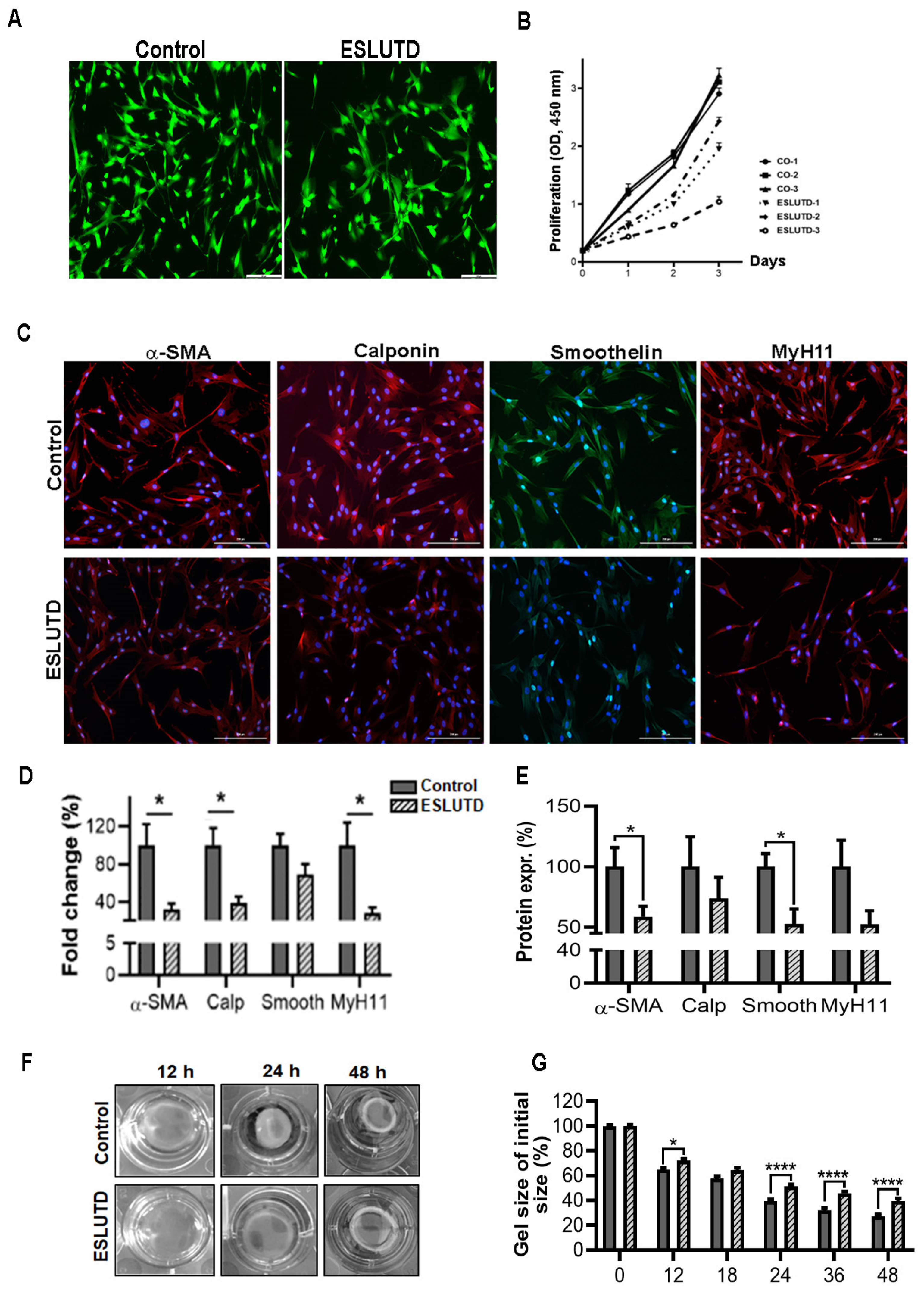
2.3. Expression of Essential Contractile Markers in Bladder-Derived Smooth Muscle Cells
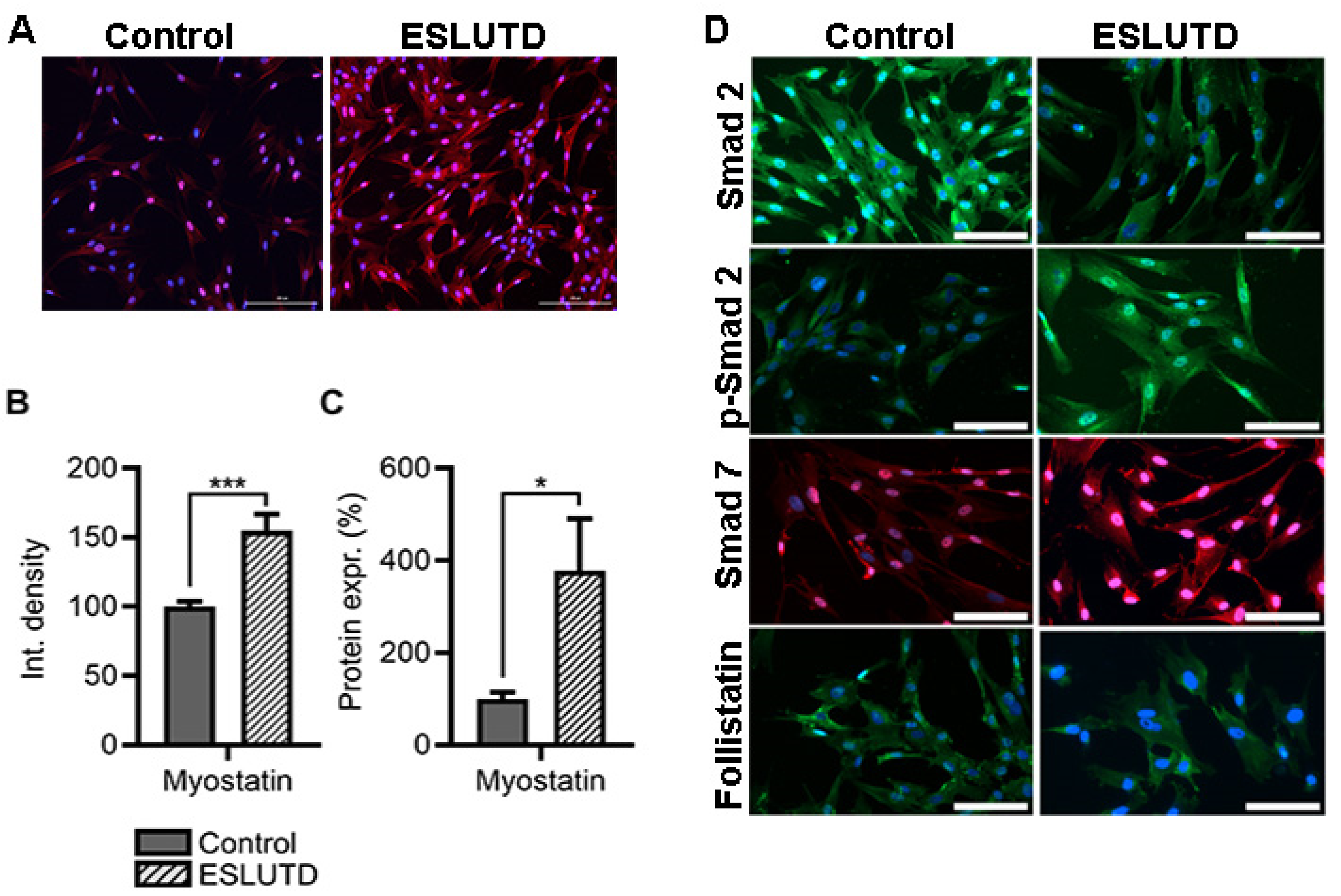
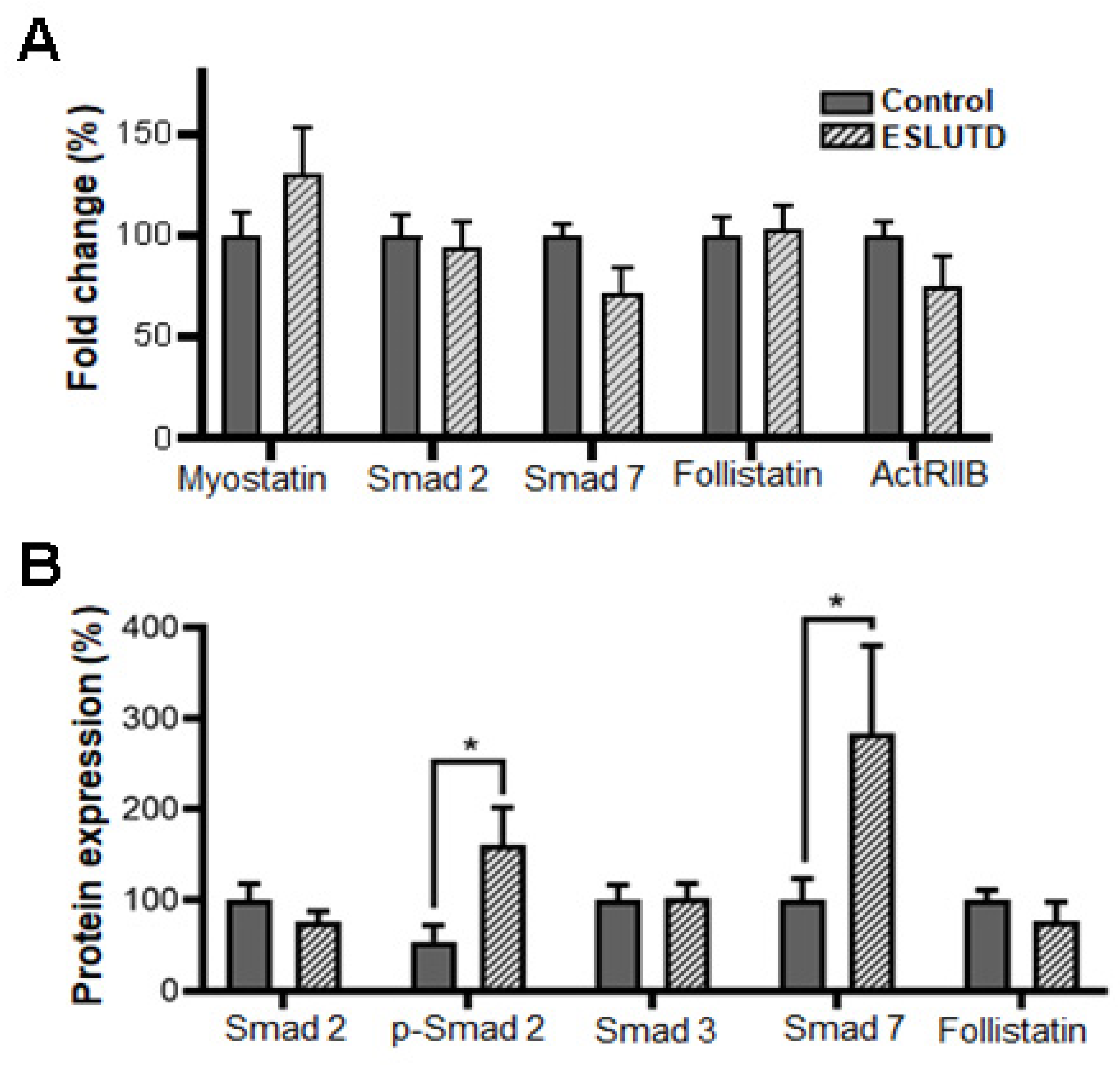
2.4. Myostatin Expression and Involvement of Smad Pathway in Bladder Smooth Muscle Cells
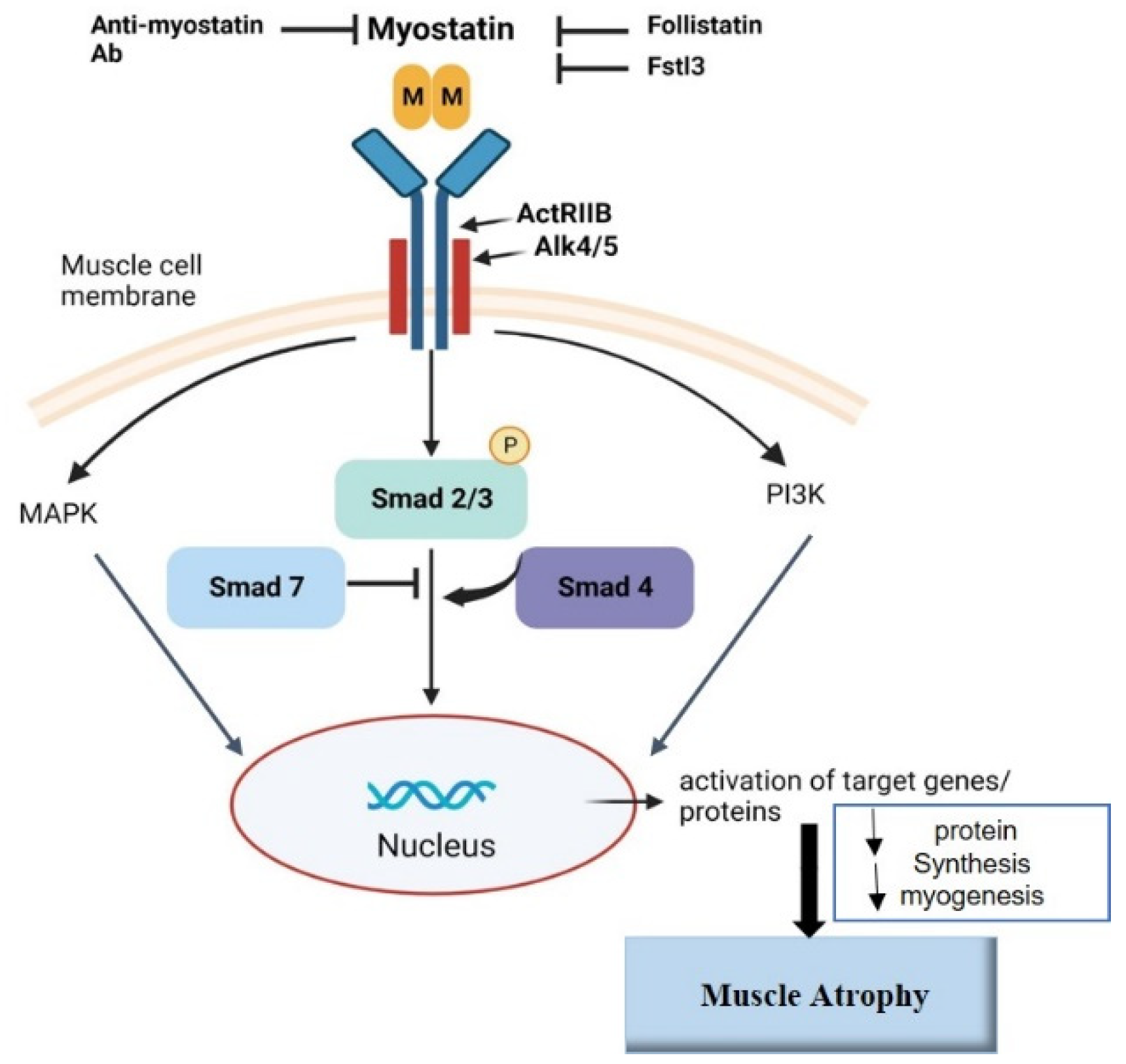
3. Discussion
4. Materials and Methods
4.1. Patient Selection and Biopsies
4.2. Isolation of Human Bladder Smooth Muscle Cells
4.3. Tissue Sample Preparation and Histological Analysis
4.4. Immunofluorescent Staining
4.5. Cell Proliferation
4.6. Quantitative Real-Time PCR
4.7. Fluorescence-Activated Cell Sorting
4.8. Immunoblotting (Automated Western Blotting—WES)
4.9. Gel Contraction Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.; Klassen, D.K.; Franz, K.M.; Giwa, S.; Lewis, J.K. Social, economic, and policy implications of organ preservation advances. Curr. Opin. Organ Transplant. 2018, 23, 336–346. [Google Scholar] [CrossRef]
- Shermadou, E.S.; Rahman, S.; Leslie, S.W. Anatomy, Abdomen and Pelvis, Bladder. In StatPearls [Internet]; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Smolar, J.; Salemi, S.; Horst, M.; Sulser, T.; Eberli, D. Stem Cells in Functional Bladder Engineering. Transfus. Med. Hemother. 2016, 43, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Horst, M.; Eberli, D.; Gobet, R.; Salemi, S. Tissue Engineering in Pediatric Bladder Reconstruction—The Road to Success. Front. Pediatr. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budzyn, J.; Trinh, H.; Raffee, S.; Atiemo, H. Bladder Augmentation (Enterocystoplasty): The Current State of a Historic Operation. Curr. Urol. Rep. 2019, 20, 50. [Google Scholar] [CrossRef]
- Kaefer, M.; Hendren, W.H.; Bauer, S.B.; Goldenblatt, P.; Peters, C.A.; Atala, A.; Retik, A.B. Reservoir Calculi: A Comparison of Reservoirs Constructed From Stomach and Other Enteric Segments. J. Urol. 1998, 160 Pt 1, 2187–2190. [Google Scholar] [CrossRef]
- Huber, A.; Badylak, S.F. Phenotypic changes in cultured smooth muscle cells: Limitation or opportunity for tissue engineering of hollow organs? J. Tissue Eng. Regen. Med. 2012, 6, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular Regulation of Contractile Smooth Muscle Cell Phenotype: Implications for Vascular Tissue Engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [Green Version]
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kambadur, R.; Matthews, K.G.; Somers, W.G.; Devlin, G.P.; Conaglen, J.V.; Fowke, P.J.; Bass, J.J. Myostatin, a transforming growth factor-beta superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 1999, 180, 1–9. [Google Scholar] [CrossRef]
- I Kovanecz, I.; Masouminia, M.; Gelfand, R.; Vernet, D.; Rajfer, J.; Gonzalez-Cadavid, N.F. Myostatin, a profibrotic factor and the main inhibitor of striated muscle mass, is present in the penile and vascular smooth muscle. Int. J. Impot. Res. 2017, 29, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Ciarmela, P.; Islam, S.; Reis, F.M.; Gray, P.C.; Bloise, E.; Petraglia, F.; Vale, W.; Castellucci, M. Growth factors and myometrium: Biological effects in uterine fibroid and possible clinical implications. Hum. Reprod. Updat. 2011, 17, 772–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef]
- Sharma, M.; McFarlane, C.; Kambadur, R.; Kukreti, H.; Bonala, S.; Srinivasan, S. Myostatin: Expanding horizons. IUBMB Life 2015, 67, 589–600. [Google Scholar] [CrossRef]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The Myostatin Propeptide and the Follistatin-related Gene Are Inhibitory Binding Proteins of Myostatin in Normal Serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef] [Green Version]
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 2004, 270, 19–30. [Google Scholar] [CrossRef]
- Inoue, S.; Orimo, A.; Hosoi, T.; Matsuse, T.; Hashimoto, M.; Yamada, R.; Ouchi, Y.; Orimo, H.; Muramatsu, M. Expression of follistatin, an activin-binding protein, in vascular smooth muscle cells and arteriosclerotic lesions. Arterioscler. Thromb. 1993, 13, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.J.; Qiu, Y.; Hewick, R.M.; Wolfman, N.M. Regulation of Myostatin in Vivo by Growth and Differentiation Factor-Associated Serum Protein-1: A Novel Protein with Protease Inhibitor and Follistatin Domains. Mol. Endocrinol. 2003, 17, 1144–1154. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.; Lin, B.K. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 2013, 7, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J.; et al. Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skelet. Muscle 2015, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersen, J.; Kjaer, M.; Schjerling, P. The possible role of myostatin in skeletal muscle atrophy and cachexia. Scand. J. Med. Sci. Sports 2006, 16, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.-R.; Chen, R.-C. Serum myostatin levels and skeletal muscle wasting in chronic obstructive pulmonary disease. Respir. Med. 2012, 106, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataille, S.; Chauveau, P.; Fouque, D.; Aparicio, M.; Koppe, L. Myostatin and muscle atrophy during chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- García, P.S.; Cabbabe, A.; Kambadur, R.; Nicholas, G.; Csete, M. Brief-reports: Elevated myostatin levels in patients with liver disease: A potential contributor to skeletal muscle wasting. Anesth. Analg. 2010, 111, 707–709. [Google Scholar] [CrossRef]
- Bish, L.T.; George, I.; Maybaum, S.; Yang, J.; Chen, J.M.; Sweeney, H.L. Myostatin Is Elevated in Congenital Heart Disease and After Mechanical Unloading. PLoS ONE 2011, 6, e23818. [Google Scholar] [CrossRef] [Green Version]
- Lokireddy, S.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; Kambadur, R.; McFarlane, C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am. J. Physiol. Physiol. 2011, 301, C1316–C1324. [Google Scholar] [CrossRef] [Green Version]
- Bdolah, Y.; Segal, A.; Tanksale, P.; Karumanchi, S.A.; Lecker, S.H. Atrophy-related ubiquitin ligases atrogin-1 and MuRF-1 are associated with uterine smooth muscle involution in the postpartum period. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R971–R976. [Google Scholar] [CrossRef] [Green Version]
- Rodino-Klapac, L.R.; Bs, A.M.H.; Kota, J.; Bs, C.H.; Kaspar, B.K.; Mendell, J.R. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 2009, 39, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Bogdanovich, S.; Krag, T.O.B.; Barton, E.R.; Morris, L.D.; Ahima, R.S.; Khurana, T.S. Myostatin (GDF8) blockage improves muscle function in MDX mouse. J. Neurol. Sci. 2002, 199, S65. [Google Scholar]
- Biesemann, N.; Mendler, L.; Kostin, S.; Wietelmann, A.; Borchardt, T.; Braun, T. Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. 2015, 361, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Deveaud, C.M.; Macarak, E.J.; Kucich, U.; Ewalt, D.H.; Abrams, W.R.; Howard, P.S. Molecular analysis of collagens in bladder fibrosis. J. Urol. 1998, 160, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Johal, N.; Cao, K.X.; Xie, B.; Millar, M.; Davda, R.; Ahmed, A.; Kanai, A.J.; Wood, D.N.; Jabr, R.I.; Fry, C.H. Contractile and Structural Properties of Detrusor from Children with Neurogenic Lower Urinary Tract Dysfunction. Biology 2021, 10, 863. [Google Scholar] [CrossRef] [PubMed]
- Johal, N.; Cao, K.; Arthurs, C.; Millar, M.; Thrasivoulou, C.; Ahmed, A.; Jabr, R.I.; Wood, D.; Cuckow, P.; Fry, C.H. Contractile function of detrusor smooth muscle from children with posterior urethral valves—The role of fibrosis. J. Pediatr. Urol. 2021, 17, 100.e1–100.e10. [Google Scholar] [CrossRef] [PubMed]
- Eberli, D.; Horst, M.; Mortezavi, A.; Andersson, K.-E.; Gobet, R.; Sulser, T.; Simon, H.-U.; Salemi, S. Increased autophagy contributes to impaired smooth muscle function in neurogenic lower urinary tract dysfunction. Neurourol. Urodyn. 2018, 37, 2414–2424. [Google Scholar] [CrossRef]
- Gonzalez-Cadavid, N.F.; Taylor, W.E.; Yarasheski, K.; Sinha-Hikim, I.; Ma, K.; Ezzat, S.; Shen, R.; Lalani, R.; Asa, S.; Mamita, M.; et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc. Natl. Acad. Sci. USA 1998, 95, 14938–14943. [Google Scholar] [CrossRef] [Green Version]
- Gruson, D.; Ahn, S.A.; Ketelslegers, J.; Rousseau, M.F. Increased plasma myostatin in heart failure. Eur. J. Heart Fail. 2011, 13, 734–736. [Google Scholar] [CrossRef]
- Verzola, D.; Barisione, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef]
- Biesemann, N.; Mendler, L.; Wietelmann, A.; Hermann, S.; Schäfers, M.; Krüger, M.; Boettger, T.; Borchardt, T.; Braun, T. Myostatin Regulates Energy Homeostasis in the Heart and Prevents Heart Failure. Circ. Res. 2014, 115, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Fang, L.; Cong, L.; Chung, J.P.W.; Li, T.C.; Chan, D.Y.L. Myostatin: A multifunctional role in human female reproduction and fertility—A short review. Reprod. Biol. Endocrinol. 2022, 20, 96. [Google Scholar] [CrossRef] [PubMed]
- Lieber, R.L.; Ward, S.R. Cellular Mechanisms of Tissue Fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol. 2013, 305, C241–C252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Zaidy, S.A.; Sahenk, Z.; Rodino-Klapac, L.R.; Kaspar, B.; Mendell, J.R. Follistatin Gene Therapy Improves Ambulation in Becker Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anaya-Segura, M.A.; García-Martínez, F.A.; Montes-Almanza, L.; Díaz, B.-G.; Ávila-Ramírez, G.; Alvarez-Maya, I.; Coral-Vázquez, R.M.; Mondragón-Terán, P.; Escobar-Cedillo, R.E.; García-Calderón, N.; et al. Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection. Molecules 2015, 20, 11154–11172. [Google Scholar] [CrossRef]
- Lach-Trifilieff, E.; Minetti, G.C.; Sheppard, K.; Ibebunjo, C.; Feige, J.N.; Hartmann, S.; Brachat, S.; Rivet, H.; Koelbing, C.; Morvan, F.; et al. An Antibody Blocking Activin Type II Receptors Induces Strong Skeletal Muscle Hypertrophy and Protects from Atrophy. Mol. Cell. Biol. 2014, 34, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- St. Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet. Muscle 2017, 7, 1–12. [Google Scholar]
- Lin, H.-K.; Cowan, R.; Moore, P.; Zhang, Y.; Yang, Q.; Peterson, J.A., Jr.; Tomasek, J.J.; Kropp, B.P.; Cheng, E.Y. Characterization of Neuropathic Bladder Smooth Muscle Cells in Culture. J. Urol. 2004, 171, 1348–1352. [Google Scholar] [CrossRef]
- Pokrywczynska, M.; Balcerczyk, D.; Jundzill, A.; Gagat, M.; Czapiewska, M.; Kloskowski, T.; Nowacki, M.; Gastecka, A.M.; Bodnar, M.; Grzanka, A.; et al. Isolation, expansion and characterization of porcine urinary bladder smooth muscle cells for tissue engineering. Biol. Proced. Online 2016, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Ngo, P.; Ramalingam, P.; Phillips, J.A.; Furuta, G.T. Collagen Gel Contraction Assay. Methods Mol. Biol. 2006, 341, 103–109. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salemi, S.; Schori, L.J.; Gerwinn, T.; Horst, M.; Eberli, D. Myostatin Overexpression and Smad Pathway in Detrusor Derived from Pediatric Patients with End-Stage Lower Urinary Tract Dysfunction. Int. J. Mol. Sci. 2023, 24, 4462. https://doi.org/10.3390/ijms24054462
Salemi S, Schori LJ, Gerwinn T, Horst M, Eberli D. Myostatin Overexpression and Smad Pathway in Detrusor Derived from Pediatric Patients with End-Stage Lower Urinary Tract Dysfunction. International Journal of Molecular Sciences. 2023; 24(5):4462. https://doi.org/10.3390/ijms24054462
Chicago/Turabian StyleSalemi, Souzan, Larissa J. Schori, Tim Gerwinn, Maya Horst, and Daniel Eberli. 2023. "Myostatin Overexpression and Smad Pathway in Detrusor Derived from Pediatric Patients with End-Stage Lower Urinary Tract Dysfunction" International Journal of Molecular Sciences 24, no. 5: 4462. https://doi.org/10.3390/ijms24054462