
Aducanumab—Hope or Disappointment for Alzheimer’s Disease

Abstract
:1. Introduction
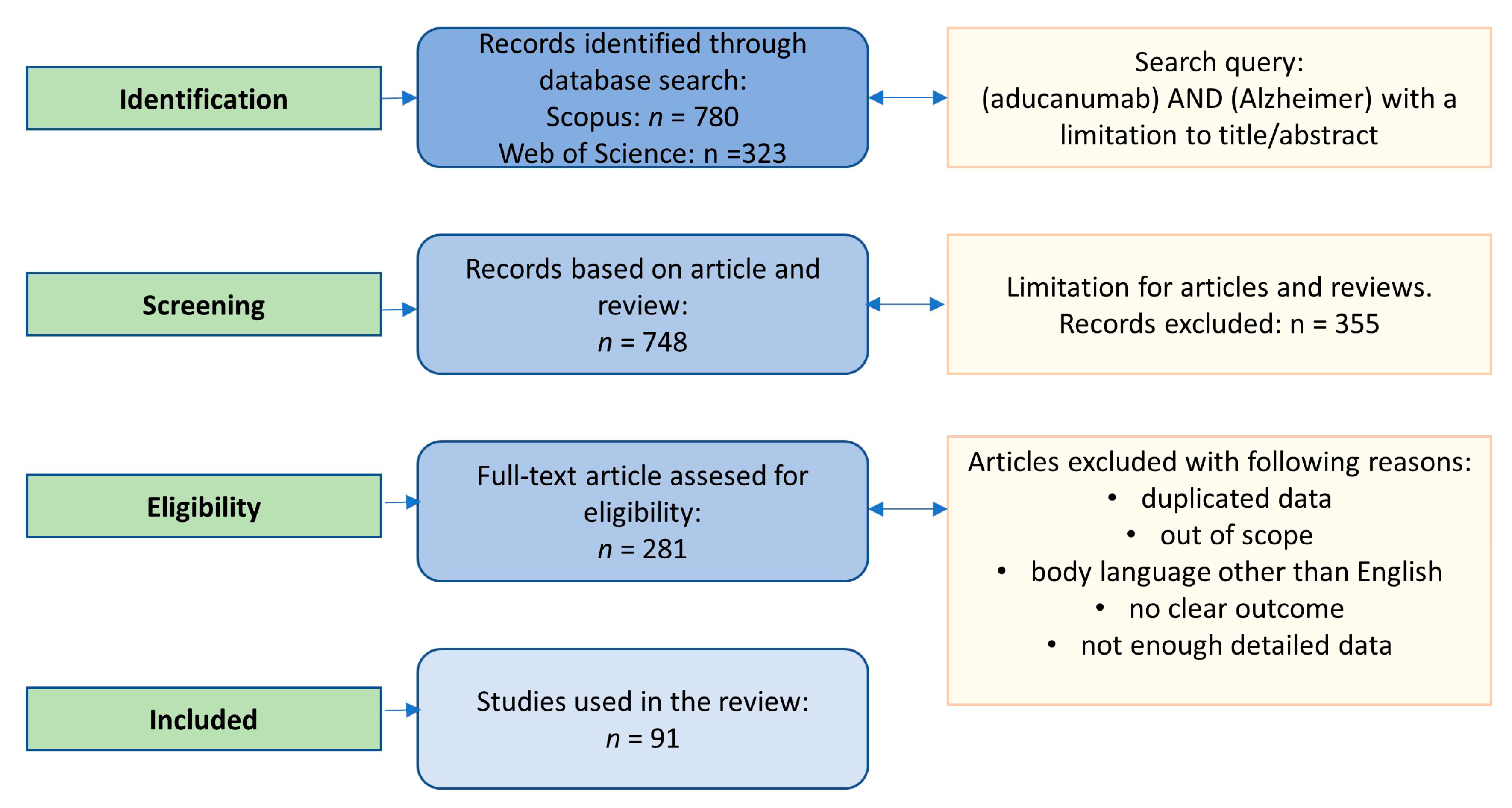
2. Search Strategy
3. Introduction to Amyloid Beta
- Binding by hydrophobic interaction (engaging three hydrophobic groups: Val18-Ala21, Lys28, and Val40-Ala42), which can affect cell viability (Figure 3).
- Encouraging the production of more amyloid fiber by releasing two short fragments able to replicate.
- Enhancing amyloid production by mitochondrial involvement, which can lead to dysfunction of ER and mitochondria.
- Forming new Aβ oligomers, which can generate higher-toxicity effects.
- Aβ is always a feature of AD, but NFTs are not.
- Amyloids under elevated concentration are a neurotoxin in tissue culture.
- Fibrillar amyloid beta can induce mitogen-activated protein kinase, leading to tau phosphorylation and the formation of neurofibrillary tangles.
- An increase in the level of Aβ is associated with mutations in the APP, PS1, and PS2 genes.
- In vivo studies based on transgenic mice revealed a correlation between the Aβ concentration and amyloid plaques [47].
- Treatment of Aβ leads to NFT clearance in the early stages [48].
- Aβ oligomer levels per plaque are much lower than in AD brains, which indicates that plaques can sequester oligomers in a non-diffusible, less neurotoxic state [49].
- The physiological concentration of Aβ does not play a neurotoxic role in the organism [50].
- Cell death is not caused by the presence of only amyloid plaques, whereas the presence of tau is always associated with neurodegeneration [32].
- Moreover:
- Clinical studies did not always indicate a correlation between the presence of plaques and AD [51].
- Lowering amyloid levels through immunotherapy against the amyloid caused harm to the recipients, including neuroinflammation [52].
- Reduction of Aβ levels did not impact behavioral changes (water-maze and Y-maze tests with transgenic animals) [53].
- Individual differences exist in the ability of inflammatory cells to effectively clear senile plaques in the brain [51].
- Individual variations in brain plasticity and the ability to restore brain function after an injury have been noted [54].
- The neurotoxic properties of amyloid oligomers precede the less neurotoxic senile plaques and could very likely be the main cause of cognitive impairment [55].
4. Monoclonal Antibodies in AD Therapy
- Murine antibodies: Procured entirely from mouse proteins, they are recognized as allogeneic proteins, hence leading to polyclonal human anti-mouse antibody (HAMA) reactions, usually 2–3 weeks after their initial infusion [56]. Currently, the antibodies are not used in neurology.
- Chimeric antibodies: The characteristic feature of the antibodies is the fact that they contain only 34% mouse proteins in variable regions of the antibody. This has an impact on the lower incidence of the HAMA reaction in comparison to murine mAbs. Additional advantages are the longer half-life and increased affinity for the antigen, which creates better pharmacodynamic and pharmacokinetic profiles. In neurology, only rituximab and infliximab are used [59,60].
- Humanized antibodies: The antibodies are 90% human and 10% mouse protein. In this case, they are less immunogenic and acquire biological functions, along with retaining the specificity and binding affinity of the ‘parental’ murine mAbs [61]. These antibodies are commonly used in neurological indications.
- Fully Human Monoclonal Antibodies: New technologies and transgenic mice allowed the production of 100% human mAbs. The antibodies are characterized by the complete removal of murine components. This has led to fewer immunogenic reactions, as well as better pharmacokinetic profiles. Today, fully human mAbs are used in migraine and multiple sclerosis therapy (erenumab and ofatumumab, respectively) [58].
5. Aducanumab–Positive and Negative Sides of Therapy
5.1. Phase 1b: PRIME
5.2. Phase 3: ENGAGE and EMERGE
6. Negative Aspects of Aducanumab Approvement
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer Disease, a Multifactorial Disorder Seeking Multi-Therapies. Alzheimer’s Dement. 2010, 6, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef]
- EClinicalMedicine Alzheimer’s Disease: Still in Need of a Cure! eClinicalMedicine 2021, 39. [CrossRef]
- Delbreil, P.; Rabanel, J.-M.; Banquy, X.; Brambilla, D. Therapeutic Nanotechnologies for Alzheimer’s Disease: A Critical Analysis of Recent Trends and Findings. Adv. Drug Deliv. Rev. 2022, 187, 114397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Wojtunik-Kulesza, K.; Oniszczuk, A.; Waksmundzka-hajnos, M. An Attempt to Elucidate the Role of Iron and Zinc Ions in Development of Alzheimer’s and Parkinson’s Diseases. Biomed. Pharmacother. = Biomed. Pharmacother. 2019, 111, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef] [PubMed]
- Nehra, G.; Bauer, B.; Hartz, A.M.S. Blood-Brain Barrier Leakage in Alzheimer’s Disease: From Discovery to Clinical Relevance. Pharmacol. Ther. 2022, 234, 108119. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- The Road to Precision Medicine: Eliminating the “One Size Fits” Approach in Alzheimer’s Disease|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S0753332222007260?token=7271AC75FE6E94A4083968D35ED1989EEE59FC4FA9C399BB5783392DBB8B37BBE68B9D0ABCE4D240E36096116DCE184E&originRegion=eu-west-1&originCreation=20230208093757 (accessed on 8 February 2023).
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Berkowitz, C.L.; Mosconi, L.; Scheyer, O.; Rahman, A.; Hristov, H.; Isaacson, R.S. Precision Medicine for Alzheimer’s Disease Prevention. Healthcare 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s Disease-Related Gray Matter Atrophy by B-Vitamin Treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, M.; Zhang, J.; Chen, S.; Huang, Y.; Chen, W.; He, L.; Zhang, Y. Role of Calcium Homeostasis in Alzheimer’s Disease. Neuropsychiatr Dis. Treat. 2022, 18, 487–498. [Google Scholar] [CrossRef]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef] [Green Version]
- Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium Dysregulation and Neuroinflammation: Discrete and Integrated Mechanisms for Age-Related Synaptic Dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, Present and Future of Therapeutic Strategies against Amyloid-β Peptides in Alzheimer’s Disease: A Systematic Review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, J.; Li, X.; Ma, L.; Hou, M.; Zhou, H.; Zhou, R. Based on Molecular Structures: Amyloid-β Generation, Clearance, Toxicity and Therapeutic Strategies. Front. Mol. Neurosci. 2022, 15, 927530. [Google Scholar] [CrossRef]
- Al Khashali, H.; Ray, R.; Coleman, K.-L.; Atali, S.; Haddad, B.; Wareham, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of the Soluble Amyloid Precursor Protein α (SAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. Int. J. Mol. Sci. 2022, 23, 10746. [Google Scholar] [CrossRef]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedrós, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the Advances in Treatment for Alzheimer Disease: Strategies for Combating β-Amyloid Protein. Neurología 2018, 33, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.L.; Gray, M.D.; Sopher, B.L.; Hu, Q.; Hearn, M.G.; Pham, D.G.; Dinulos, M.B.; Fukuchi, K.-I.; Sisodia, S.S.; Miller, M.A.; et al. CDNA Cloning and Chromosome Mapping of the Human Fe65 Gene: Interaction of the Conserved Cytoplasmic Domains of the Human β-Amyloid Precursor Protein and Its Homologues with the Mouse Fe65 Protein. Hum. Mol. Genet. 1996, 5, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Ribarič, S. Peptides as Potential Therapeutics for Alzheimer’s Disease. Molecules 2018, 23, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, L.; Brimijoin, S.; Vaux, D.J. In Vivo Localization of Human Acetylcholinesterase-Derived Species in a β-Sheet Conformation at the Core of Senile Plaques in Alzheimer’s Disease. J. Biol. Chem. 2019, 294, 6253–6272. [Google Scholar] [CrossRef]
- Sarroukh, R.; Cerf, E.; Derclaye, S.; Dufrêne, Y.F.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. Transformation of Amyloid β(1–40) Oligomers into Fibrils Is Characterized by a Major Change in Secondary Structure. Cell. Mol. Life Sci. 2011, 68, 1429–1438. [Google Scholar] [CrossRef]
- Santoro, A.; Grimaldi, M.; Buonocore, M.; Stillitano, I.; Gloria, A.; Santin, M.; Bobba, F.; Sublimi Saponetti, M.; Ciaglia, E.; D’Ursi, A.M. New Aβ(1-42) Ligands from Anti-Amyloid Antibodies: Design, Synthesis, and Structural Interaction. Eur. J. Med. Chem. 2022, 237, 114400. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Glabe, C.G. Distinct Early Folding and Aggregation Properties of Alzheimer Amyloid-β Peptides Aβ40 and Aβ42: STABLE TRIMER OR TETRAMER FORMATION BY Aβ42*. J. Biol. Chem. 2006, 281, 24414–24422. [Google Scholar] [CrossRef] [Green Version]
- Sriram, S.; Mehkri, Y.; Quintin, S.; Lucke-Wold, B. Shared Pathophysiology: Understanding Stroke and Alzheimer’s Disease. Clin. Neurol. Neurosurg. 2022, 218, 107306. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Galanis, C.; Fellenz, M.; Becker, D.; Bold, C.; Lichtenthaler, S.F.; Müller, U.C.; Deller, T.; Vlachos, A. Amyloid-Beta Mediates Homeostatic Synaptic Plasticity. J. Neurosci. 2021, 41, 5157–5172. [Google Scholar] [CrossRef]
- Styr, B.; Slutsky, I. Imbalance between Firing Homeostasis and Synaptic Plasticity Drives Early-Phase Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 463–473. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Li, J.; Du, P.; Jin, W.; Gao, G.; Cui, D. Senile Plaques in Alzheimer’s Disease Arise from Aβ- and Cathepsin D-Enriched Mixtures Leaking out during Intravascular Haemolysis and Microaneurysm Rupture. FEBS Lett. 2022. [Google Scholar] [CrossRef]
- Dierksen, G.A.; Skehan, M.E.; Khan, M.A.; Jeng, J.; Nandigam, R.K.; Becker, J.A.; Kumar, A.; Neal, K.L.; Betensky, R.A.; Frosch, M.P.; et al. Spatial Relation between Microbleeds and Amyloid Deposits in Amyloid Angiopathy. Ann. Neurol. 2010, 68, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Cullen, K.; Kócsi, Z.; Stone, J. Pericapillary Haem-Rich Deposits: Evidence for Microhaemorrhages in Aging Human Cerebral Cortex. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2006, 25, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The Systemic Iron-Regulatory Proteins Hepcidin and Ferroportin Are Reduced in the Brain in Alzheimer’s Disease. Acta Neuropathol. Commun. 2013, 1, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Lendel, C. Extracellular Protein Components of Amyloid Plaques and Their Roles in Alzheimer’s Disease Pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Casey, J. Cerebral Microhemorrhage Implicated in Progression of Alzheimer’s Disease. Nat. Rev. Neurol. 2006, 2, 234. [Google Scholar] [CrossRef]
- Cordonnier, C.; van der Flier, W.M. Brain Microbleeds and Alzheimer’s Disease: Innocent Observation or Key Player? Brain 2011, 134, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Weiner, R.L.; Dumitrescu, L.; Hohman, T.J. Protective Genes and Pathways in Alzheimer’s Disease: Moving towards Precision Interventions. Mol. Neurodegener. 2021, 16, 29. [Google Scholar] [CrossRef]
- Sepulveda-Falla, D.; Chavez-Gutierrez, L.; Portelius, E.; Vélez, J.I.; Dujardin, S.; Barrera-Ocampo, A.; Dinkel, F.; Hagel, C.; Puig, B.; Mastronardi, C.; et al. A Multifactorial Model of Pathology for Age of Onset Heterogeneity in Familial Alzheimer’s Disease. Acta Neuropathol. 2021, 141, 217–233. [Google Scholar] [CrossRef]
- Van der Linden, M.; Juillerat Van der Linden, A.-C. A Life-Course and Multifactorial Approach to Alzheimer’s Disease: Implications for Research, Clinical Assessment and Intervention Practices. Dementia 2018, 17, 880–895. [Google Scholar] [CrossRef] [Green Version]
- Berson, A.; Knobloch, M.; Hanan, M.; Diamant, S.; Sharoni, M.; Schuppli, D.; Geyer, B.C.; Ravid, R.; Mor, T.S.; Nitsch, R.M.; et al. Changes in Readthrough Acetylcholinesterase Expression Modulate Amyloid-Beta Pathology. Brain 2008, 131, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Rees, T.M.; Berson, A.; Sklan, E.H.; Younkin, L.; Younkin, S.; Brimijoin, S.; Soreq, H. Memory Deficits Correlating with Acetylcholinesterase Splice Shift and Amyloid Burden in Doubly Transgenic Mice. Curr. Alzheimer Res. 2005, 2, 291–300. [Google Scholar] [CrossRef]
- Luo, J.J.; Wallace, W.; Kusiak, J.W. A Tough Trek in the Development of an Anti-Amyloid Therapy for Alzheimer’s Disease: Do We See Hope in the Distance? J. Neurol. Sci. 2022, 438, 120294. [Google Scholar] [CrossRef]
- Takeuchi, A.; Irizarry, M.C.; Duff, K.; Saido, T.C.; Hsiao Ashe, K.; Hasegawa, M.; Mann, D.M.A.; Hyman, B.T.; Iwatsubo, T. Age-Related Amyloid β Deposition in Transgenic Mice Overexpressing Both Alzheimer Mutant Presenilin 1 and Amyloid β Precursor Protein Swedish Mutant Is Not Associated with Global Neuronal Loss. Am. J. Pathol. 2000, 157, 331–339. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Aβ Immunotherapy Leads to Clearance of Early, but Not Late, Hyperphosphorylated Tau Aggregates via the Proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-Beta Oligomerization in Alzheimer Dementia versus High-Pathology Controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. Deposits of Fibrillar Aβ Do Not Cause Neuronal Loss or Ferritin Expression in Adult Rat Brain. J. Neural Transm. 2003, 110, 381–400. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-Term Effects of Aβ42 Immunisation in Alzheimer’s Disease: Follow-up of a Randomised, Placebo-Controlled Phase I Trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Kshirsagar, S.; Alvir, R.V.; Hindle, A.; Kumar, S.; Vijayan, M.; Pradeepkiran, J.A.; Reddy, A.P.; Ramasubramanian, B.; Reddy, P.H. Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease. Cells 2022, 11, 733. [Google Scholar] [CrossRef]
- Plasticity in the Working Memory System: Life Span Changes and Response to Injury–Sean Froudist-Walsh, Diana López-Barroso, María José Torres-Prioris, Paula L. Croxson, Marcelo L. Berthier. 2018. Available online: https://journals.sagepub.com/doi/10.1177/1073858417717210 (accessed on 9 February 2023).
- Sciaccaluga, M.; Megaro, A.; Bellomo, G.; Ruffolo, G.; Romoli, M.; Palma, E.; Costa, C. An Unbalanced Synaptic Transmission: Cause or Consequence of the Amyloid Oligomers Neurotoxicity? Int. J. Mol. Sci. 2021, 22, 5991. [Google Scholar] [CrossRef]
- Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals 2021, 14, 92. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Kung, P.C.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal Antibodies Defining Distinctive Human T Cell Surface Antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT: Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Steinitz, M. Three Decades of Human Monoclonal Antibodies: Past, Present and Future Developments. Hum. Antibodies 2009, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Buss, N.A.P.S.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal Antibody Therapeutics: History and Future. Curr. Opin. Pharm. 2012, 12, 615–622. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Lemere, C.A. Immunotherapy for Alzheimer’s Disease: Hoops and Hurdles. Mol. Neurodegener. 2013, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.; Choo, P.C.; Waragai, M.; Inoue, S.; Masliah, E.; Hashimoto, M. Reconsideration of Alzheimer’s Disease Therapy from a Viewpoint of Amyloidogenic Evolvability. J. Alzheimer’s Dis. Rep. 2022, 6, 207–210. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56, reprinted in Nature 2017, 546, 564. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer Disease and Aducanumab: Adjusting Our Approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and Tolerability of BAN2401--a Clinical Study in Alzheimer’s Disease with a Protofibril Selective Aβ Antibody. Alzheimer’s Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Donanemab|ALZFORUM. Available online: https://www.alzforum.org/therapeutics/donanemab (accessed on 16 January 2023).
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A Phase III Randomized Trial of Gantenerumab in Prodromal Alzheimer’s Disease. Alzheimer’s Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Gantenerumab|ALZFORUM. Available online: https://www.alzforum.org/therapeutics/gantenerumab (accessed on 16 January 2023).
- Vaz, M.; Silvestre, S. Alzheimer’s Disease: Recent Treatment Strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Gosuranemab|ALZFORUM. Available online: https://www.alzforum.org/therapeutics/gosuranemab (accessed on 16 January 2023).
- Semorinemab|ALZFORUM. Available online: https://www.alzforum.org/therapeutics/semorinemab (accessed on 16 January 2023).
- Tilavonemab|ALZFORUM. Available online: https://www.alzforum.org/therapeutics/tilavonemab (accessed on 16 January 2023).
- Coerver, K.; Yu, M.M.; D’Abreu, A.; Wasserman, M.; Nair, K.V. Practical Considerations in the Administration of Aducanumab for the Neurologist. Neurol. Clin. Pract. 2022, 12, 169–175. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and Kinetic Basis for the Selectivity of Aducanumab for Aggregated Forms of Amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [Green Version]
- Haddad, H.W.; Malone, G.W.; Comardelle, N.J.; Degueure, A.E.; Kaye, A.M.; Kaye, A.D. Aducanumab, a Novel Anti-Amyloid Monoclonal Antibody, for the Treatment of Alzheimer’s Disease: A Comprehensive Review. Health Psychol. Res. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Bennett, D.A. Aducanumab and the “Post-Amyloid” Era of Alzheimer Research? Neuron 2021, 109, 3045–3047. [Google Scholar] [CrossRef]
- Terao, I.; Honyashiki, M.; Inoue, T. Comparative Efficacy of Lithium and Aducanumab for Cognitive Decline in Patients with Mild Cognitive Impairment or Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Ageing Res. Rev. 2022, 81, 101709. [Google Scholar] [CrossRef]
- Biogen, A. Phase 3 Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Aducanumab (BIIB037) in Subjects with Early Alzheimer’s Disease; 2021. Available online: clinicaltrials.gov (accessed on 15 January 2023).
- Day, G.S.; Scarmeas, N.; Dubinsky, R.; Coerver, K.; Mostacero, A.; West, B.; Wessels, S.R.; Armstrong, M.J. Aducanumab Use in Symptomatic Alzheimer Disease Evidence in Focus: A Report of the AAN Guidelines Subcommittee. Neurology 2022, 98, 619–631. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A Plaque-Specific Antibody Clears Existing β-Amyloid Plaques in Alzheimer’s Disease Mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Sharma, N.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Felemban, S.G.; Alsubayiel, A.M.; et al. “Aducanumab” Making a Comeback in Alzheimer’s Disease: An Old Wine in a New Bottle. Biomed. Pharm. 2022, 148, 112746. [Google Scholar] [CrossRef]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic Fingerprints Differentiate the Mechanisms of Action of Anti-Aβ Antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133. [Google Scholar] [CrossRef]
- Alexander, G.C.; Karlawish, J. The Problem of Aducanumab for the Treatment of Alzheimer Disease. Ann. Intern. Med. 2021, 174, 1303–1304. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Gatsonis, C.; Apgar, C.; Chaudhary, K.; Gareen, I.; Hanna, L.; Hendrix, J.; Hillner, B.E.; Olson, C.; Lesman-Segev, O.H.; et al. Association of Amyloid Positron Emission Tomography With Subsequent Change in Clinical Management Among Medicare Beneficiaries With Mild Cognitive Impairment or Dementia. JAMA 2019, 321, 1286–1294. [Google Scholar] [CrossRef]
- Barenholtz Levy, H. Accelerated Approval of Aducanumab: Where Do We Stand Now? Ann. Pharm. 2022, 56, 736–739. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Action | Stage of Development | Advancement of Alzheimer’s Disease | References |
|---|---|---|---|---|---|
| Aducanumab | Fully human IgG1 | Against Aβ aggregation | Accepted by FDA | Prodromal to mild | [66,67] |
| Donanemab | Humanized IgG1 | Binding aggregated Aβ forms | In phase III | Mild | [68,69] |
| Gantenerumab | Fully human IgG1 | Binding aggregated Aβ forms | In two phase III Trials, The company stopped all trials in order to prepare a new Gantenerumab formula. | Prodromal to mild | [70,71] |
| Gosuranemab | Humanized IgG4 | Targeting abnormal forms of tau protein or soluble oligomers | Negative results in phase II | Prodromal to mild | [72,73] |
| Semorinemab | Humanized IgG4 | Targeting all isoforms of tau protein | A phase 3 decision is pending | Prodromal to mild | [72,74] |
| Tilavonemab | Humanized IgG4 | Targeting abnormal extracellular forms of tau protein | Trial development was stopped after phase II (2020) | Prodromal to mild | [72,75] |
| Study Name/Identification | Number Enrolled | Key Inclusion Criteria | Level of Evidence Statement |
|---|---|---|---|
| Single ascending dose study of BIIB037 in participants with AD | 53 | Clinically confirmed AD, age: 55–85 years old, others: Good health, reliable informant or caregiver | Single dose of aducanumab (up to 30 mg/kg) was safe and tolerable |
| PRIME (Multiple dose study of aducanumab) | 197 | Prodromal or mild AD, Age: 50–90 years old; others: Good health, reliable informant or caregiver | Decreasing amyloid value studied with the use of PET SUVR at 1 year vs. placebo (dosage: 3–10 mg/kg) |
| ENGAGE (Phase 3 Study) | 1647 | MCI due to AD or mild AD; Age: 50–85 years old; MMSE 24–30; others: Positive amyloid PET scan, stable doses of drugs treating AD symptoms, reliable informant or caregiver | Aducanumab (3–10 mg/kg) did not significantly affect mean change in CDR-SB scores vs. placebo over 78 weeks whereas the same doses caused decrease in amyloid PET SUVR at 78 weeks vs. placebo |
| EMERGE (Phase 3 study) | 1638 | MCI due to AD or mild AD; Age: 50–85 years old; MMSE 24–30; others: Positive amyloid PET scan, stable doses of drugs treating AD symptoms, reliable informant or caregiver | Aducanumab at a dose of 10 mg/kg results in less worsening of the CDR-SB vs. placebo at 78 weeks; degree less than a clinically relevant change; doses of 3–10 mg/kg caused decrease in amyloid PET SUVR at 78 weeks vs. placebo |
| EVOLVE | 52 | MCI due to AD or mild AD; Age: 50–85 years old; MMSE 24–30 others: Positive amyloid PET scan | NA |
| PROPEL (Single and multiple ascending dose study in Japanese participants with AD) | 21 | Clinical diagnosis of mild-moderate AD; age: 55–85 years old; others: Good health, reliable informant or caregiver | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtunik-Kulesza, K.; Rudkowska, M.; Orzeł-Sajdłowska, A. Aducanumab—Hope or Disappointment for Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4367. https://doi.org/10.3390/ijms24054367
Wojtunik-Kulesza K, Rudkowska M, Orzeł-Sajdłowska A. Aducanumab—Hope or Disappointment for Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(5):4367. https://doi.org/10.3390/ijms24054367
Chicago/Turabian StyleWojtunik-Kulesza, Karolina, Monika Rudkowska, and Anna Orzeł-Sajdłowska. 2023. "Aducanumab—Hope or Disappointment for Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 5: 4367. https://doi.org/10.3390/ijms24054367