
Vx-809, a CFTR Corrector, Acts through a General Mechanism of Protein Folding and on the Inflammatory Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
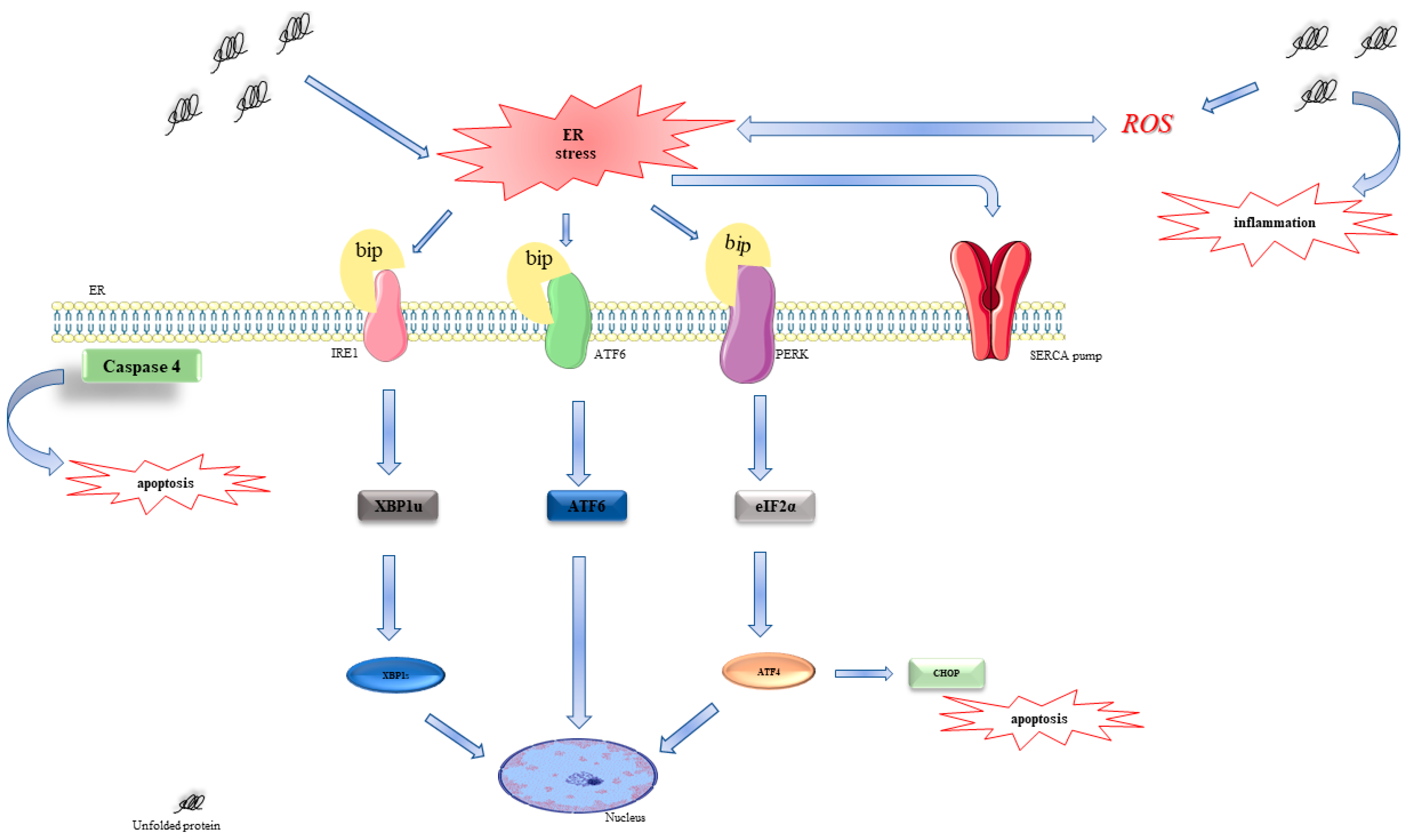
:1. Introduction
2. Results
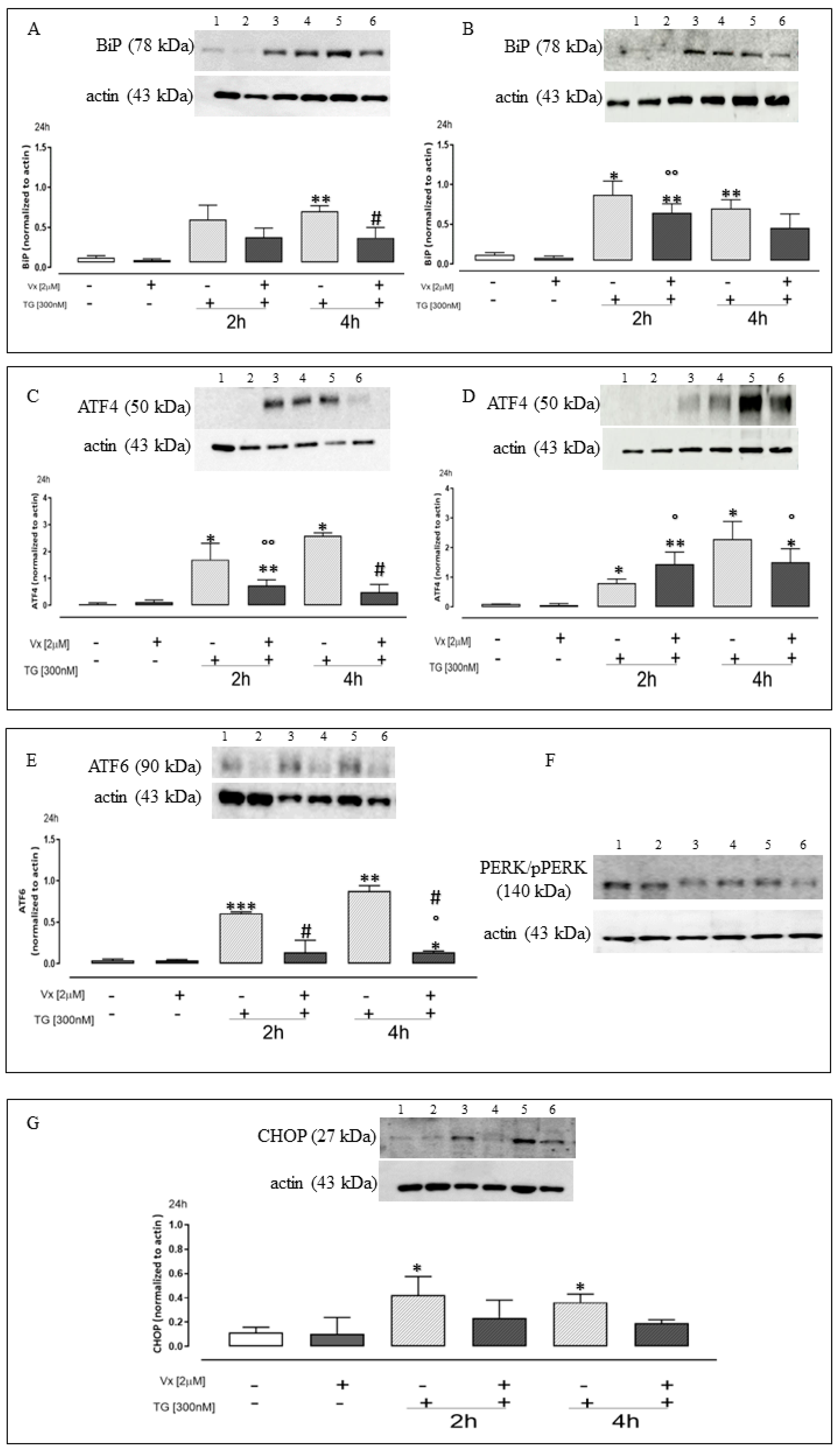
2.1. Vx-809 Interferes with UPR Activation
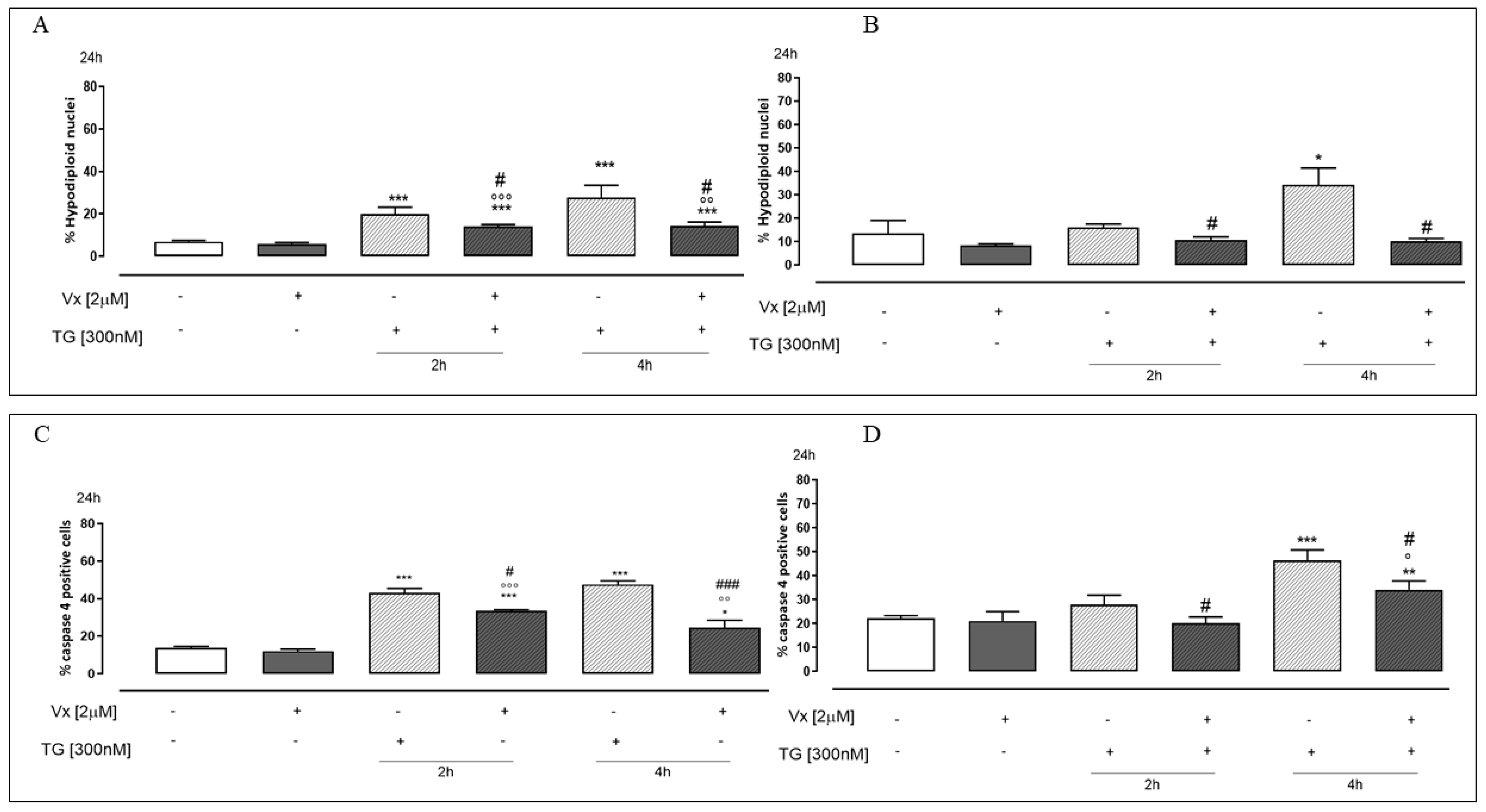
2.2. Vx-809 Counteract Thapsigargin-Induced Apoptotic Response
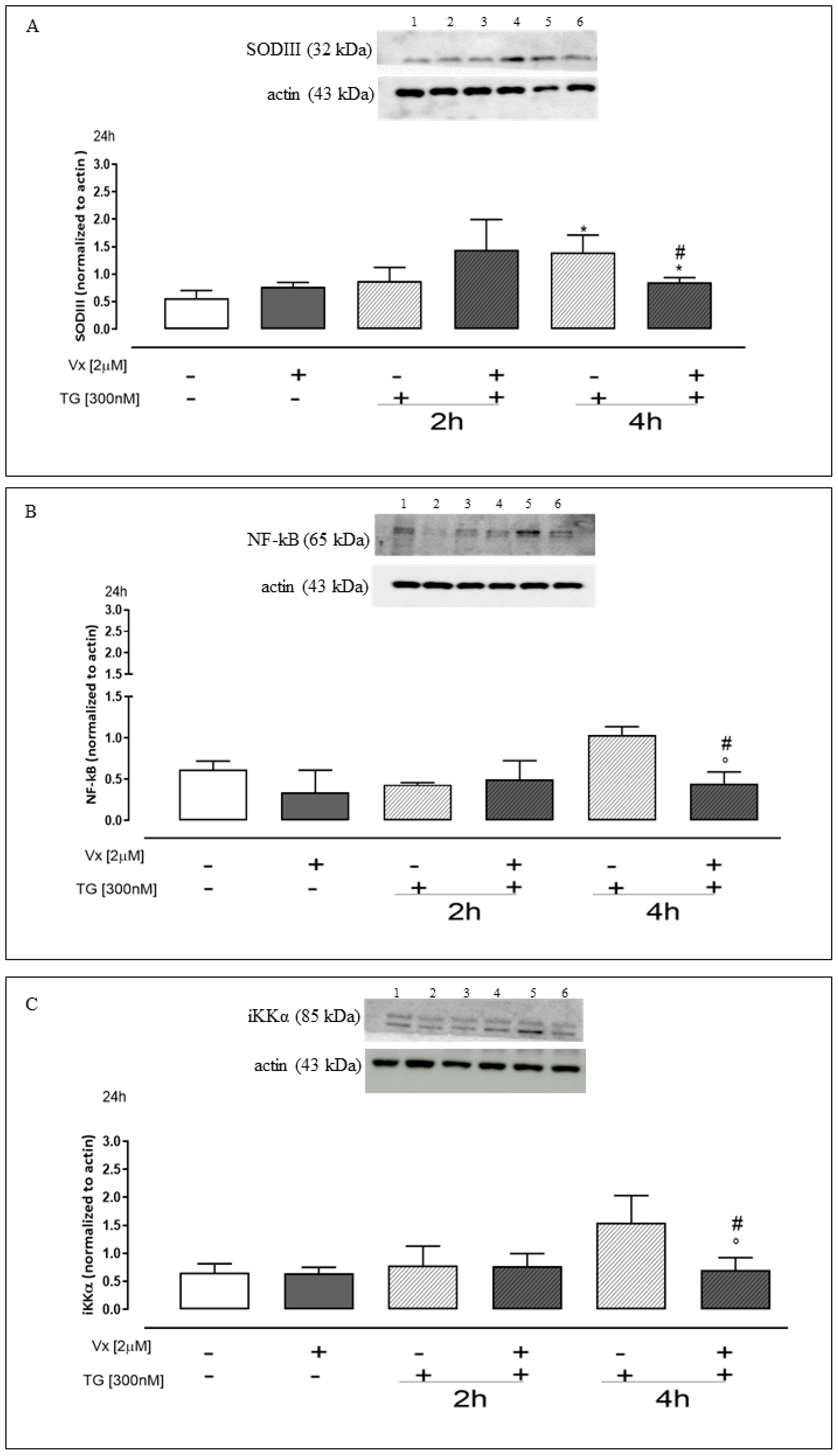
2.3. Vx-809 Is Involved in Intracellular and Mitochondrial ROS Production
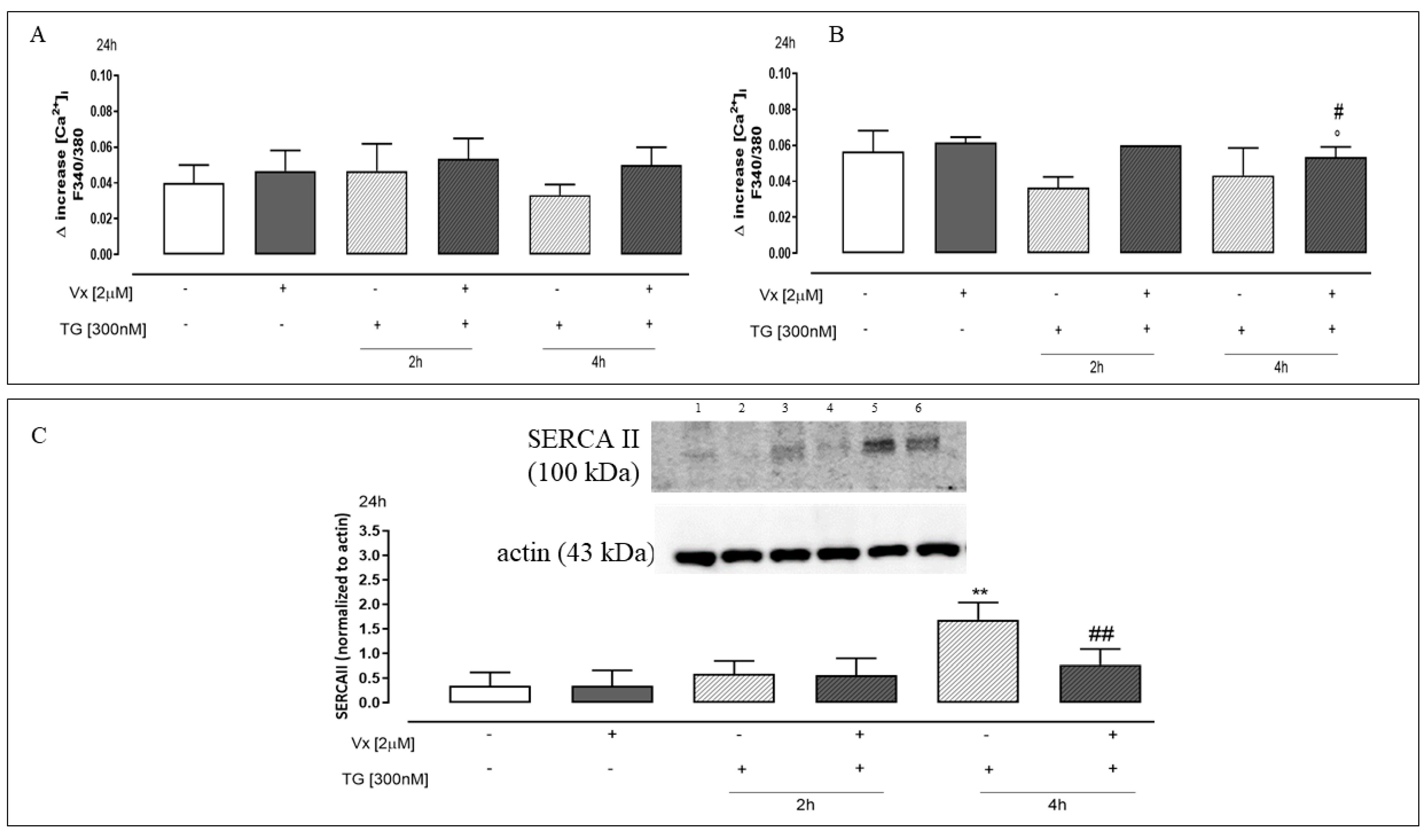
2.4. The “Corrector” Vx-809 Reduces Thapsigargin-Induced Ca2+ Homeostasis Dysregulation
2.5. Effect of Vx-809 on the Inflammatory Pathway
3. Discussion
4. Materials and Methods
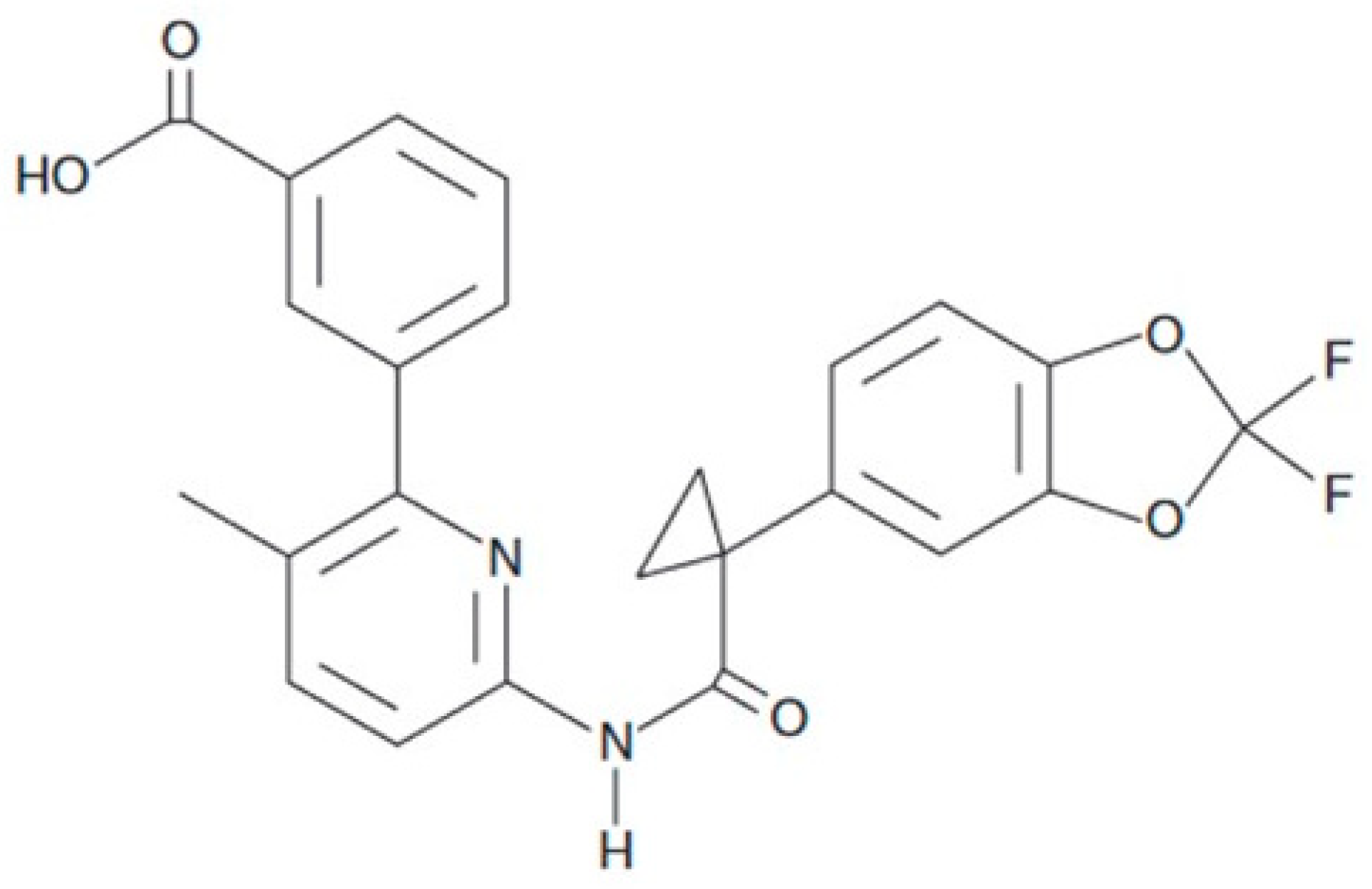
4.1. Reagents
4.2. Cell Culture
4.3. Experimental Protocol
4.4. Protein Extraction and Western Blot Analysis
4.5. Measurement of Intracellular Calcium Signaling
4.6. Intracellular and Mitochondrial ROS Release Measurement
4.7. Flow Cytometry Analysis
4.8. Determination of Hypodiploid DNA
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer’s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Gonzalez, I.; Soto, C. Misfolded protein aggregates: Mechanisms, structures and potential for disease transmission. Semin. Cell. Dev. Biol. 2011, 22, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Amodio, G.; Margarucci, L.; Moltedo, O.; Casapullo, A.; Remondelli, P. Identification of Cysteine Ubiquitylation Sites on the Sec23A Protein of the COPII Complex Required for Vesicle Formation from the ER. Open Biochem. J. 2017, 11, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lee, J.E.; Kang, J.W.; Shin, H.Y.; Lee, J.B.; Jin, D.I. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR) in Mammalian Oocyte Maturation and Preimplantation Embryo Development. Int. J. Mol. Sci. 2019, 20, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER Stress and Disease: Toward Prevention and Treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell. Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Abraham, T.; Pin, C.L.; Watson, A.J. Embryo collection induces transient activation of XBP1 arm of the ER stress response while embryo vitrification does not. Mol. Hum. Reprod. 2012, 18, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Benhamron, S.; Hadar, R.; Iwawaky, T.; So, J.S.; Lee, A.H.; Tirosh, B. Regulated IRE1-dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur. J. Immunol. 2014, 4, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef] [PubMed]
- Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. [Google Scholar] [CrossRef] [Green Version]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Pecoraro, M.; Franceschelli, S.; Pascale, M. Lumacaftor and Matrine: Possible Therapeutic Combination to Counteract the Inflammatory Process in Cystic Fibrosis. Biomolecules 2021, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER Stress in Vivo and in Vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression during Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [Green Version]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef] [Green Version]
- Eletto, D.; Boyle, S.; Argon, Y. PDIA6 regulates insulin secretion by selectively inhibiting the RIDD activity of IRE1. FASEB J. 2016, 30, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 13, 673. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The Endoplasmic Reticulum and Calcium Homeostasis in Pancreatic Beta Cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef]
- An, M.Y.; Lee, S.R.; Hwang, H.J.; Yoon, J.G.; Lee, H.J.; Cho, J.A. Antioxidant and Anti-Inflammatory Effects of Korean Black Ginseng Extract through ER Stress Pathway. Antioxidants 2021, 10, 62. [Google Scholar] [CrossRef]
- Valdivieso, Á.G.; Dugour, A.V.; Sotomayor, V.; Clauzure, M.; Figueroa, J.M.; Santa-Coloma, T.A. N-acetyl cysteine reverts the proinflammatory state induced by cigarette smoke extract in lung Calu-3 cells. Redox Biol. 2018, 16, 294–302. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef] [Green Version]
- Berkers, G.; van der Meer, R.; Heijerman, H.; Beekman, J.M.; Boj, S.F.; Vries, R.G.J.; van Mourik, P.; Doyle, J.R.; Audhya, P.; Yuan, Z.J.; et al. Lumacaftor/ivacaftor in people with cystic fibrosis with an A455E-CFTR mutation. J. Cyst. Fibros. 2021, 20, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, D.Z.; Xu, H.; Li, Y.; Wang, W.; Liu, B.L.; Zhang, L.Y. Thapsigargin induces apoptosis by impairing cytoskeleton dynamics in human lung adenocarcinoma cells. Sci. World J. 2014, 2014, 619050. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Franceschelli, S.; Bruno, A.P.; Festa, M.; Falco, A.; Gionti, E.; d’Avenia, M.; De Marco, M.; Basile, A.; Iorio, V.; Marzullo, L.; et al. BAG3 Protein Is Involved in Endothelial Cell Response to Phenethyl Isothiocyanate. Oxid. Med. Cell. Longev. 2018, 2018, 5967890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrizzo, A.; Moltedo, O.; Damato, A.; Martinello, K.; Di Pietro, P.; Oliveti, M.; Acernese, F.; Giugliano, G.; Izzo, R.; Sommella, E.; et al. New Nutraceutical Combination Reduces Blood Pressure and Improves Exercise Capacity in Hypertensive Patients via a Nitric Oxide-Dependent Mechanism. J. Am. Heart Assoc. 2020, 9, e014923. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Marzocco, S.; Franceschelli, S.; Popolo, A. Trastuzumab and Doxorubicin Sequential Administration Increases Oxidative Stress and Phosphorylation of Connexin 43 on Ser368. Int. J. Mol. Sci. 2022, 23, 6375. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Fiji, N.; Roy, D.; Andrew, M.G.D.; Shihabudeen, M.S.; Chattopadhyay, D.; Thirumurugan, K. A rapid method to assess reactive oxygen species in yeast using H2DCF-DA. Anal. Methods 2015, 7, 8572–8575. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecoraro, M.; Serra, A.; Pascale, M.; Franceschelli, S. Vx-809, a CFTR Corrector, Acts through a General Mechanism of Protein Folding and on the Inflammatory Process. Int. J. Mol. Sci. 2023, 24, 4252. https://doi.org/10.3390/ijms24044252
Pecoraro M, Serra A, Pascale M, Franceschelli S. Vx-809, a CFTR Corrector, Acts through a General Mechanism of Protein Folding and on the Inflammatory Process. International Journal of Molecular Sciences. 2023; 24(4):4252. https://doi.org/10.3390/ijms24044252
Chicago/Turabian StylePecoraro, Michela, Adele Serra, Maria Pascale, and Silvia Franceschelli. 2023. "Vx-809, a CFTR Corrector, Acts through a General Mechanism of Protein Folding and on the Inflammatory Process" International Journal of Molecular Sciences 24, no. 4: 4252. https://doi.org/10.3390/ijms24044252