
Expression and Function of BMP and Activin Membrane-Bound Inhibitor (BAMBI) in Chronic Liver Diseases and Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TGF-β in Liver Fibrosis and Tumorigenesis
3. TGF-β Family Members and Receptors in Liver Fibrosis
4. TGF-β Pathway in Inflammation
5. Cell-Type Specific Expression of BAMBI in the Liver
6. Regulation of BAMBI Expression in HSCs
7. Regulation of BAMBI by Adiponectin and Metformin
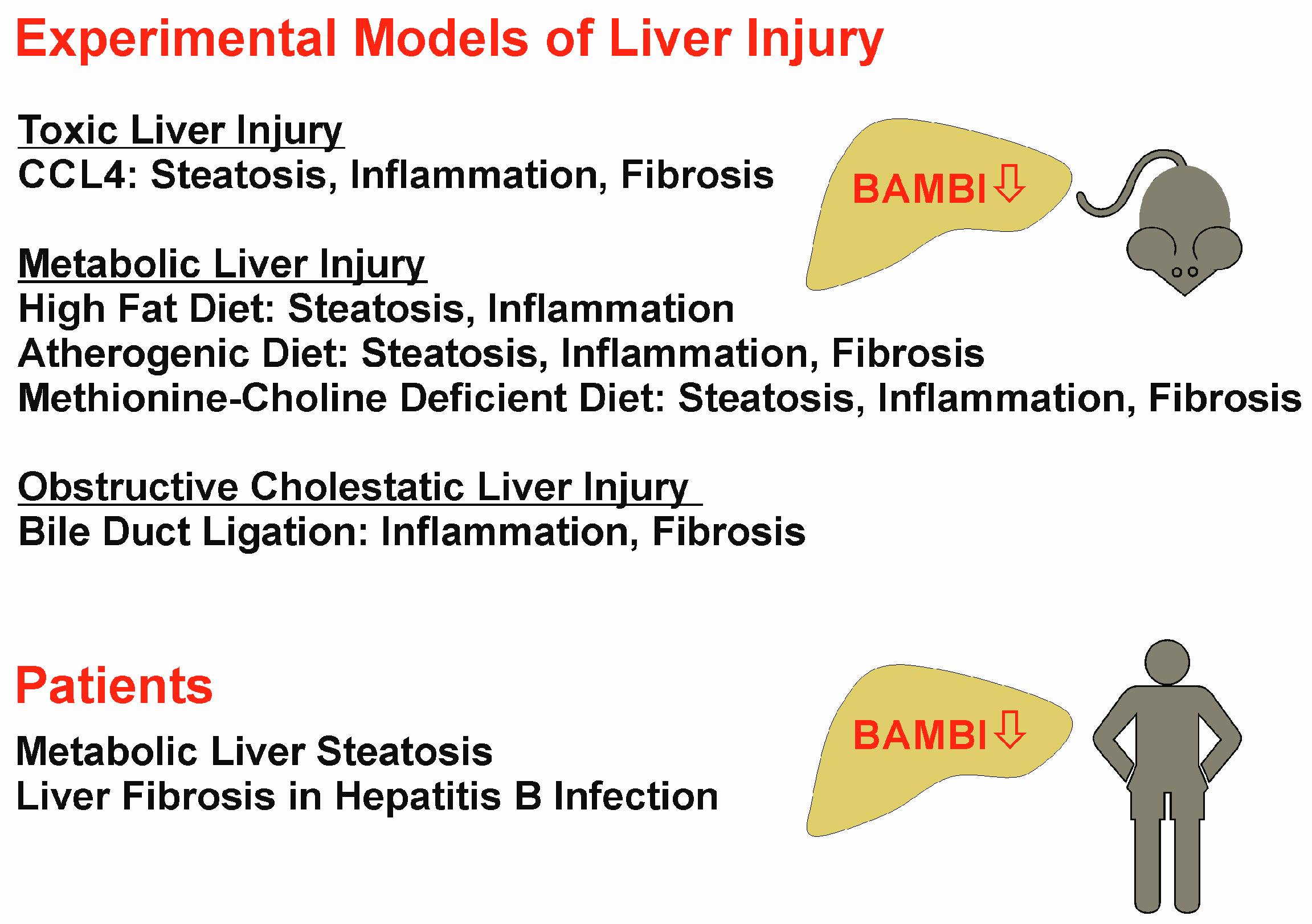
8. BAMBI Expression in Non-Alcoholic Fatty Liver Disease
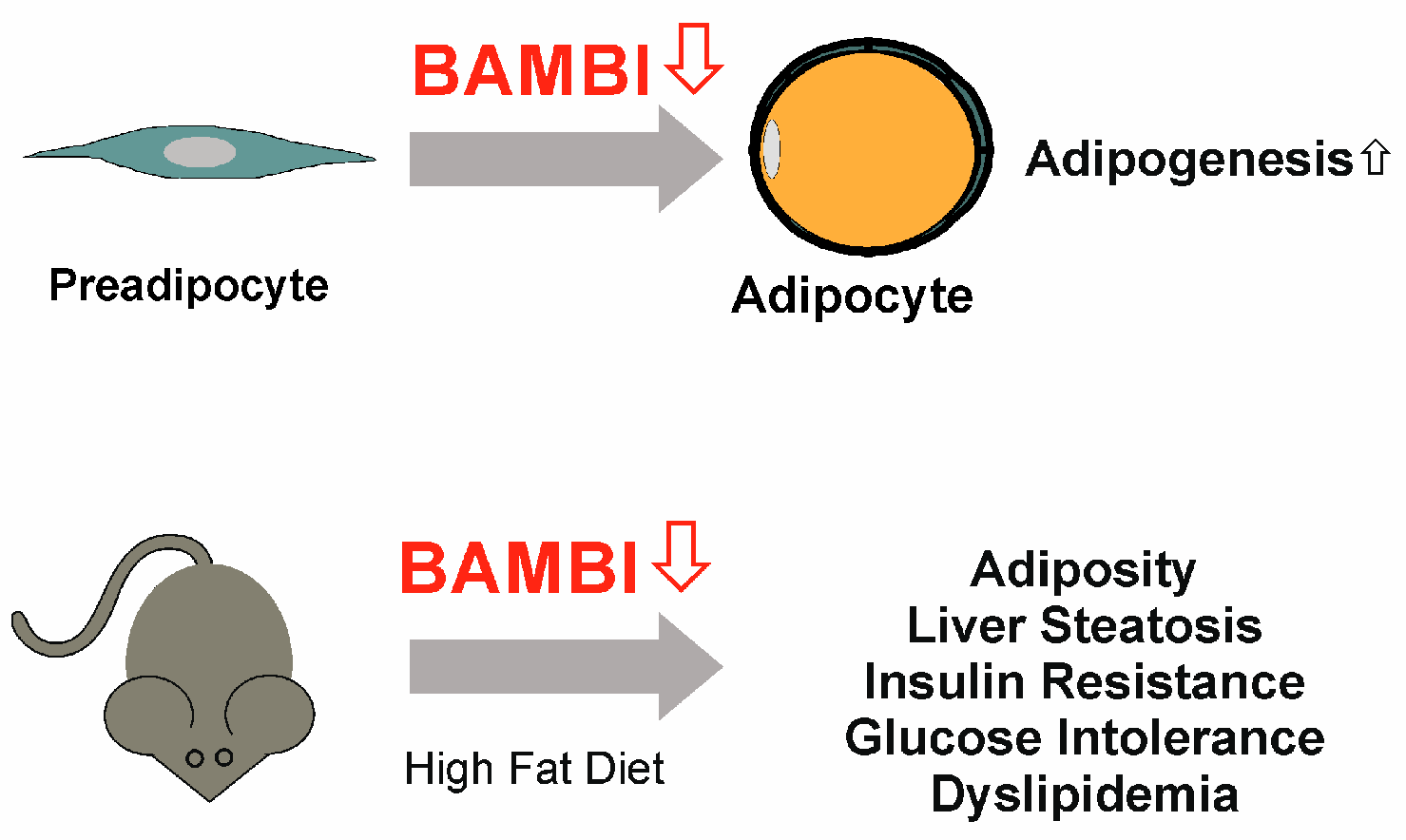
9. BAMBI Is Expressed in Adipocytes
10. BAMBI and the Wnt/β-Catenin Pathway
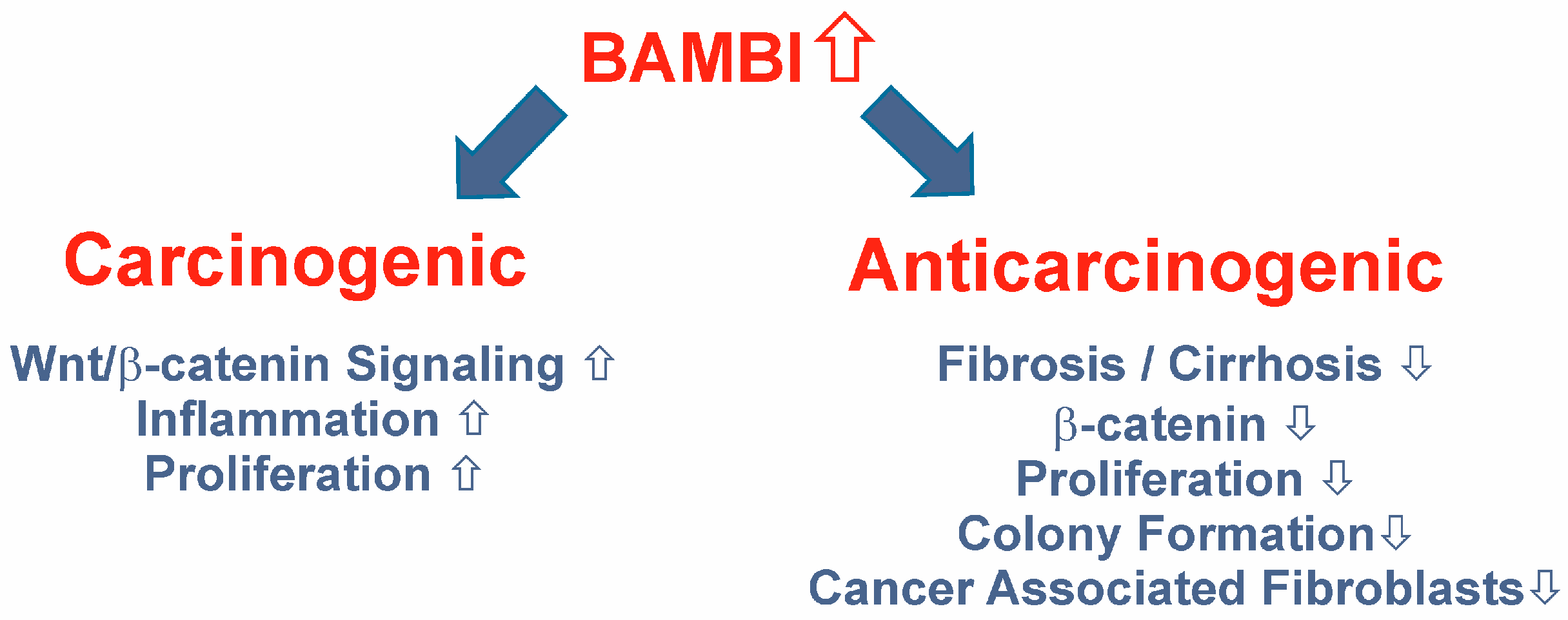
11. BAMBI Expression in HCC
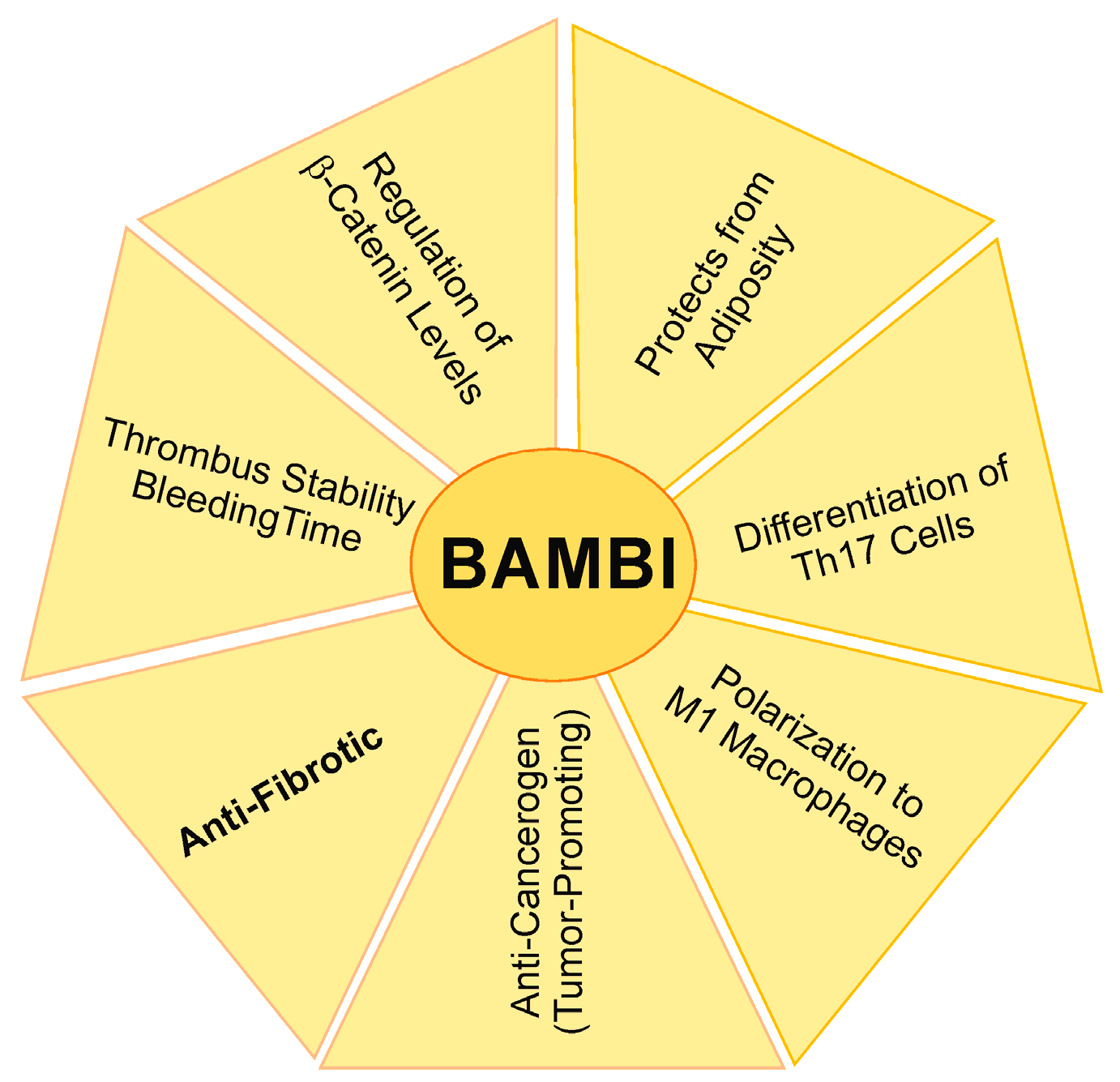
12. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, F.; Chen, Y.G. Regulation of TGF-beta receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-beta family action. Cytokine Growth Factor Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
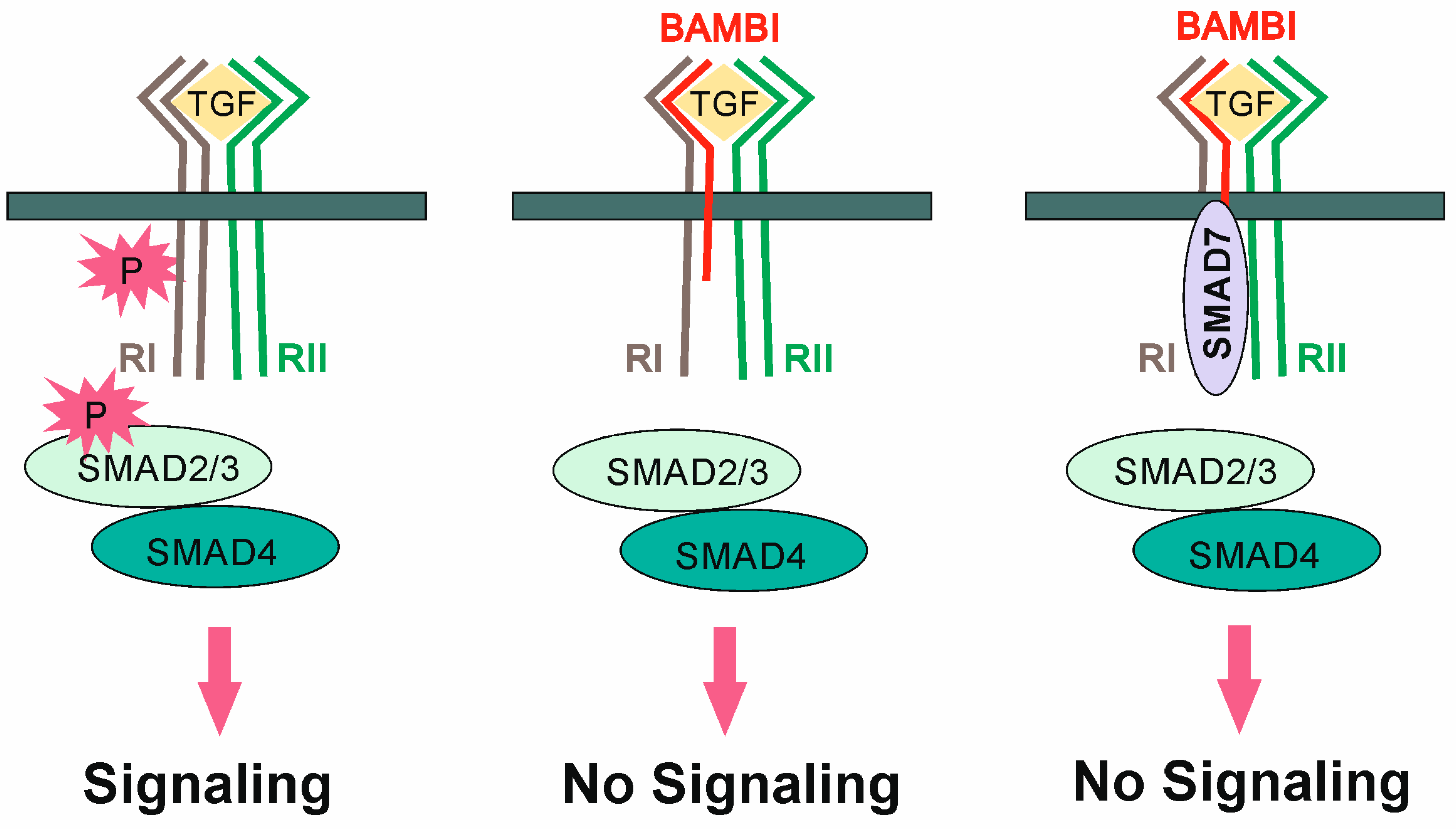
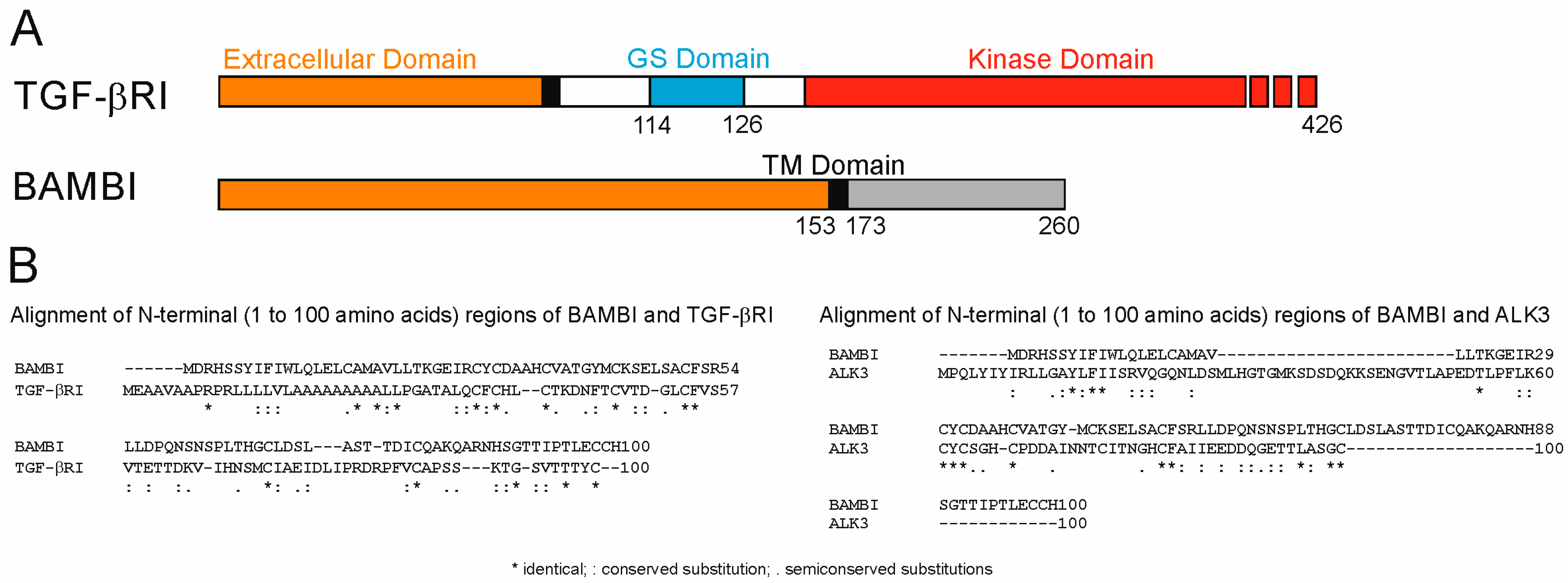
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Chen, J.; Bush, J.O.; Ovitt, C.E.; Lan, Y.; Jiang, R. The TGF-beta pseudoreceptor gene Bambi is dispensable for mouse embryonic development and postnatal survival. Genesis 2007, 45, 482–486. [Google Scholar] [CrossRef]
- Crawley JT, B.; Zalli, A.; Monkman, J.H.; Petri, A.; Lane, D.A.; Ahnstrom, J.; Salles, C., II. Defective fibrin deposition and thrombus stability in Bambi(-/-) mice are mediated by elevated anticoagulant function. J. Thromb. Haemost. 2019, 17, 1935–1949. [Google Scholar] [CrossRef]
- Salles, C., II; Monkman, J.H.; Ahnstrom, J.; Lane, D.A.; Crawley, J.T. Vessel wall BAMBI contributes to hemostasis and thrombus stability. Blood 2014, 123, 2873–2881. [Google Scholar] [CrossRef]
- Villar, A.V.; Garcia, R.; Llano, M.; Cobo, M.; Merino, D.; Lantero, A.; Tramullas, M.; Hurle, J.M.; Hurle, M.A.; Nistal, J.F. BAMBI (BMP and activin membrane-bound inhibitor) protects the murine heart from pressure-overload biomechanical stress by restraining TGF-beta signaling. Biochim. Biophys. Acta 2013, 1832, 323–335. [Google Scholar] [CrossRef]
- Degen, W.G.; Weterman, M.A.; van Groningen, J.J.; Cornelissen, I.M.; Lemmers, J.P.; Agterbos, M.A.; Geurts van Kessel, A.; Swart, G.W.; Bloemers, H.P. Expression of nma, a novel gene, inversely correlates with the metastatic potential of human melanoma cell lines and xenografts. Int. J. Cancer 1996, 65, 460–465. [Google Scholar] [CrossRef]
- Staib, F.; Krupp, M.; Maass, T.; Itzel, T.; Weinmann, A.; Lee, J.S.; Schmidt, B.; Muller, M.; Thorgeirsson, S.S.; Galle, P.R.; et al. CellMinerHCC: A microarray-based expression database for hepatocellular carcinoma cell lines. Liver Int. 2014, 34, 621–631. [Google Scholar] [CrossRef]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef]
- Yan, X.; Lin, Z.; Chen, F.; Zhao, X.; Chen, H.; Ning, Y.; Chen, Y.G. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 2009, 284, 30097–30104. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Arguinchona, L.M.; Zagona-Prizio, C.; Joyce, M.E.; Chan, E.D.; Maloney, J.P. Microvascular significance of TGF-beta axis activation in COVID-19. Front. Cardiovasc. Med. 2022, 9, 1054690. [Google Scholar] [CrossRef]
- Sherman, E.J.; Mirabelli, C.; Tang, V.T.; Khan, T.G.; Leix, K.; Kennedy, A.A.; Graham, S.E.; Willer, C.J.; Tai, A.W.; Sexton, J.Z.; et al. Identification of cell type specific ACE2 modifiers by CRISPR screening. PLoS Pathog. 2022, 18, e1010377. [Google Scholar] [CrossRef]
- Wang, X.; Lei, J.; Li, Z.; Yan, L. Potential Effects of Coronaviruses on the Liver: An Update. Front. Med. 2021, 8, 651658. [Google Scholar] [CrossRef]
- Lubnow, M.; Schmidt, B.; Fleck, M.; Salzberger, B.; Muller, T.; Peschel, G.; Schneckenpointner, R.; Lange, T.; Hitzenbichler, F.; Kieninger, M.; et al. Secondary hemophagocytic lymphohistiocytosis and severe liver injury induced by hepatic SARS-CoV-2 infection unmasking Wilson’s disease: Balancing immunosuppression. Int. J. Infect. Dis. 2021, 103, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.P.; Kurzrock, R. Epstein-Barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821. [Google Scholar] [CrossRef]
- Heawchaiyaphum, C.; Pientong, C.; Yoshiyama, H.; Iizasa, H.; Panthong, W.; Ekalaksananan, T. General Features and Novel Gene Signatures That Identify Epstein-Barr Virus-Associated Epithelial Cancers. Cancers 2021, 14, 31. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Paquissi, F.C. Immunity and Fibrogenesis: The Role of Th17/IL-17 Axis in HBV and HCV-induced Chronic Hepatitis and Progression to Cirrhosis. Front. Immunol. 2017, 8, 1195. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Lewinska, M.; Andersen, J.B. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 2022, 4, 100479. [Google Scholar] [CrossRef]
- Zakhari, S. Bermuda Triangle for the liver: Alcohol, obesity, and viral hepatitis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 18–25. [Google Scholar] [CrossRef]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [Google Scholar] [CrossRef]
- Charlton, M.R.; Pockros, P.J.; Harrison, S.A. Impact of obesity on treatment of chronic hepatitis C. Hepatology 2006, 43, 1177–1186. [Google Scholar] [CrossRef]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in chronic hepatitis C: Why does it really matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef]
- Wanninger, J.; Neumeier, M.; Hellerbrand, C.; Schacherer, D.; Bauer, S.; Weiss, T.S.; Huber, H.; Schaffler, A.; Aslanidis, C.; Scholmerich, J.; et al. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim. Biophys. Acta 2011, 1811, 626–633. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.C.; Rosso, N.; Tiribelli, C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Baccante, G.; Mincione, G.; Di Febbo, C.; Coppa, A.; Angelucci, D.; Lapenna, D.; Cuccurullo, F.; Colletta, G.; Porreca, E. Increased type II transforming growth factor-beta receptor expression in liver cells during cholesterol challenge. Atherosclerosis 2000, 152, 51–57. [Google Scholar] [CrossRef]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef]
- Kirstein, M.M.; Vogel, A. The pathogenesis of hepatocellular carcinoma. Dig. Dis. 2014, 32, 545–553. [Google Scholar] [CrossRef]
- Arrese, M.; Hernandez, A.; Astete, L.; Estrada, L.; Cabello-Verrugio, C.; Cabrera, D. TGF-beta and Hepatocellular Carcinoma: When A Friend Becomes An Enemy. Curr. Protein Pept. Sci. 2018, 19, 1172–1179. [Google Scholar] [CrossRef]
- Herrera, B.; Addante, A.; Sanchez, A. BMP Signalling at the Crossroad of Liver Fibrosis and Regeneration. Int. J. Mol. Sci. 2017, 19, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Guo, D.Y.; Xu, B.; Shi, X.Y.; Li, J.T.; Duan, L.F. Study on the relationship between hepatic fibrosis and epithelial-mesenchymal transition in intrahepatic cells. Biomed. Pharmacother. 2020, 129, 110413. [Google Scholar] [CrossRef]
- Munoz-Felix, J.M.; Gonzalez-Nunez, M.; Lopez-Novoa, J.M. ALK1-Smad1/5 signaling pathway in fibrosis development: Friend or foe? Cytokine Growth Factor Rev. 2013, 24, 523–537. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The role of Snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Vaquero, J.; Fernandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-beta Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef]
- Le, V.Q.; Iacob, R.E.; Zhao, B.; Su, Y.; Tian, Y.; Toohey, C.; Engen, J.R.; Springer, T.A. Protection of the Prodomain alpha1-Helix Correlates with Latency in the Transforming Growth Factor-beta Family. J. Mol. Biol. 2022, 434, 167439. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Z.C. The role of bone morphogenetic proteins in liver fibrosis. Gastroenterol. Hepatol. 2021, 12, 17–20. [Google Scholar] [CrossRef]
- Arndt, S.; Wacker, E.; Dorn, C.; Koch, A.; Saugspier, M.; Thasler, W.E.; Hartmann, A.; Bosserhoff, A.K.; Hellerbrand, C. Enhanced expression of BMP6 inhibits hepatic fibrosis in non-alcoholic fatty liver disease. Gut 2015, 64, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.M.; Cai, M.; Lv, Y.F.; Huang, Y.H.; Li, H.H. Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Mol. Ther. 2012, 20, 2043–2051. [Google Scholar] [CrossRef]
- Bi, J.; Ge, S. Potential roles of BMP9 in liver fibrosis. Int. J. Mol. Sci. 2014, 15, 20656–20667. [Google Scholar] [CrossRef]
- Bloomer, S.A.; Brown, K.E. Hepcidin and Iron Metabolism in Experimental Liver Injury. Am. J. Pathol. 2021, 191, 1165–1179. [Google Scholar] [CrossRef]
- Bi, W.R.; Jin, C.X.; Xu, G.T.; Yang, C.Q. Bone morphogenetic protein-7 regulates Snail signaling in carbon tetrachloride-induced fibrosis in the rat liver. Exp. Ther. Med. 2012, 4, 1022–1026. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adh. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef]
- Cicchini, C.; Amicone, L.; Alonzi, T.; Marchetti, A.; Mancone, C.; Tripodi, M. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int. 2015, 35, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Wiercinska, E.; Wickert, L.; Denecke, B.; Said, H.M.; Hamzavi, J.; Gressner, A.M.; Thorikay, M.; ten Dijke, P.; Mertens, P.R.; Breitkopf, K.; et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology 2006, 43, 1032–1041. [Google Scholar] [CrossRef]
- Kiagiadaki, F.; Kampa, M.; Voumvouraki, A.; Castanas, E.; Kouroumalis, E.; Notas, G. Activin-A causes Hepatic stellate cell activation via the induction of TNFalpha and TGFbeta in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 891–899. [Google Scholar] [CrossRef]
- Yang, Y.R.; Bu, F.T.; Yang, Y.; Li, H.; Huang, C.; Meng, X.M.; Zhang, L.; Lv, X.W.; Li, J. LEFTY2 alleviates hepatic stellate cell activation and liver fibrosis by regulating the TGF-beta1/Smad3 pathway. Mol. Immunol. 2020, 126, 31–39. [Google Scholar] [CrossRef]
- Dai, Z.; Song, G.; Balakrishnan, A.; Yang, T.; Yuan, Q.; Mobus, S.; Weiss, A.C.; Bentler, M.; Zhu, J.; Jiang, X.; et al. Growth differentiation factor 11 attenuates liver fibrosis via expansion of liver progenitor cells. Gut 2020, 69, 1104–1115. [Google Scholar] [CrossRef]
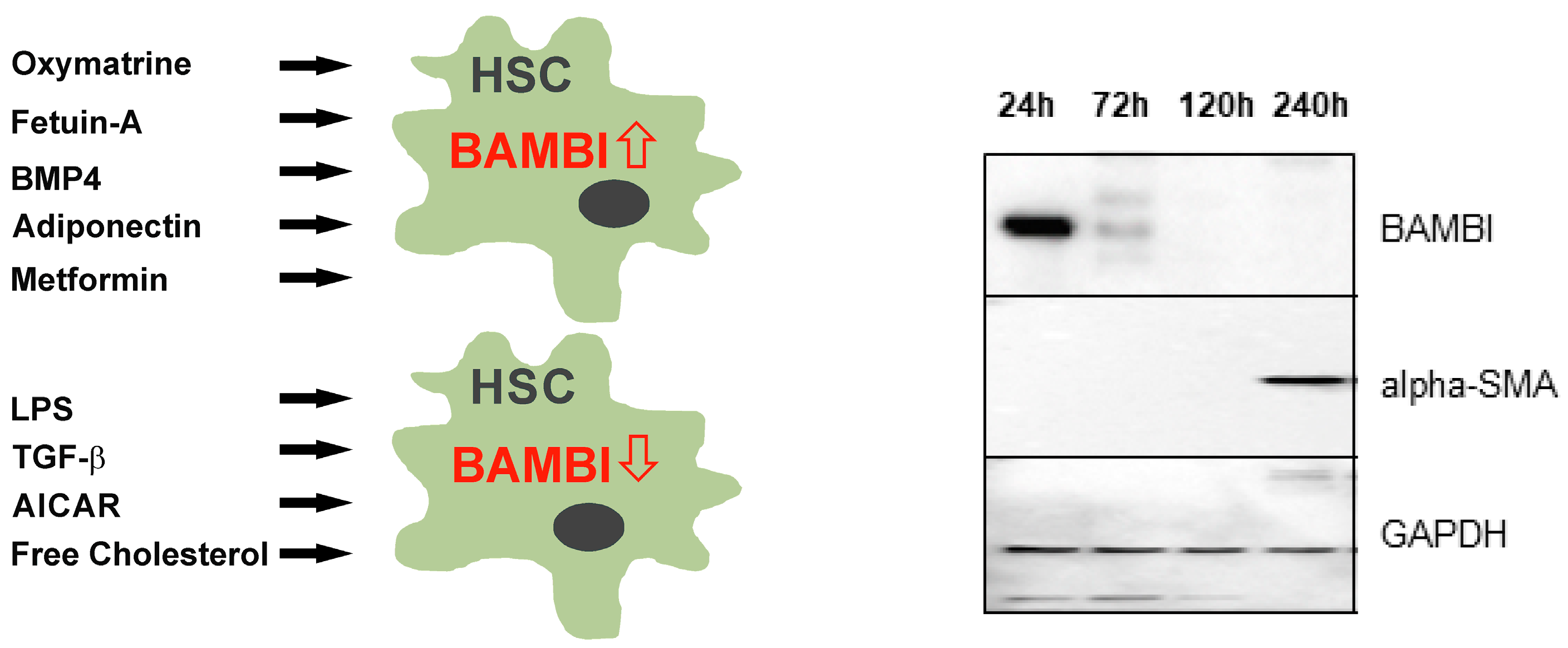
- Wanninger, J.; Neumeier, M.; Bauer, S.; Weiss, T.S.; Eisinger, K.; Walter, R.; Dorn, C.; Hellerbrand, C.; Schaffler, A.; Buechler, C. Adiponectin induces the transforming growth factor decoy receptor BAMBI in human hepatocytes. FEBS Lett. 2011, 585, 1338–1344. [Google Scholar] [CrossRef]
- Weng, H.L.; Ciuclan, L.; Liu, Y.; Hamzavi, J.; Godoy, P.; Gaitantzi, H.; Kanzler, S.; Heuchel, R.; Ueberham, U.; Gebhardt, R.; et al. Profibrogenic transforming growth factor-beta/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology 2007, 46, 1257–1270. [Google Scholar] [CrossRef]
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008, 28, 1065–1079. [Google Scholar] [CrossRef]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef]
- Leask, A.; Chen, S.; Pala, D.; Brigstock, D.R. Regulation of CCN2 mRNA expression and promoter activity in activated hepatic stellate cells. J. Cell Commun. Signal. 2008, 2, 49–56. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; Ten Dijke, P. Transforming growth factor-beta (TGF-beta)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [Green Version]
- Son, G.; Hines, I.N.; Lindquist, J.; Schrum, L.W.; Rippe, R.A. Inhibition of phosphatidylinositol 3-kinase signaling in hepatic stellate cells blocks the progression of hepatic fibrosis. Hepatology 2009, 50, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.P.; Salmon, M.; Buckley, C.D.; Adams, D.H. Immune interactions in hepatic fibrosis. Clin. Liver Dis. 2008, 12, 861–882. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Naiki, Y.; Michelsen, K.S.; Zhang, W.; Chen, S.; Doherty, T.M.; Arditi, M. Transforming growth factor-beta differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J. Biol. Chem. 2005, 280, 5491–5495. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.G.; Letterio, J.J.; Kulkarni, A.B.; Karlsson, S.; Roberts, A.B.; Sporn, M.B. Transforming growth factor beta 1 (TGF-beta 1) controls expression of major histocompatibility genes in the postnatal mouse: Aberrant histocompatibility antigen expression in the pathogenesis of the TGF-beta 1 null mouse phenotype. Proc. Natl. Acad. Sci. USA 1993, 90, 9944–9948. [Google Scholar] [CrossRef]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; Van Waes, C. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef]
- Wang, W.; Gao, W.; Zhu, Q.; Alasbahi, A.; Seki, E.; Yang, L. TAK1: A Molecular Link Between Liver Inflammation, Fibrosis, Steatosis, and Carcinogenesis. Front. Cell Dev. Biol. 2021, 9, 734749. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Kim, S.I.; Choi, M.E. TGF-beta-activated kinase-1: New insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res. Clin. Pract. 2012, 31, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Mu, C.; Zhang, Z.; He, X.; Liu, X. The Love-Hate Relationship Between TGF-beta Signaling and the Immune System During Development and Tumorigenesis. Front. Immunol. 2022, 13, 891268. [Google Scholar] [CrossRef]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef]
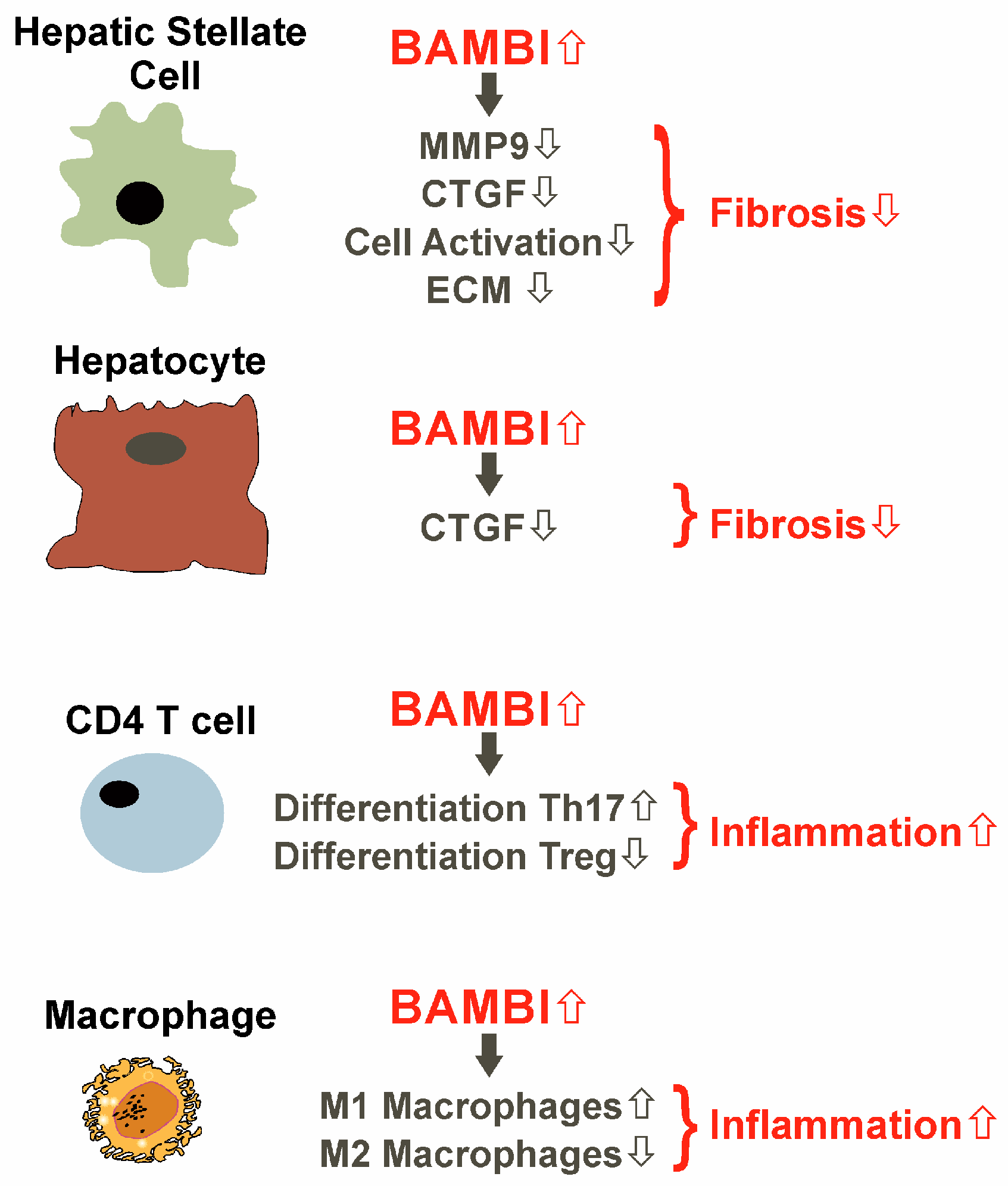
- Postigo, J.; Iglesias, M.; Alvarez, P.; Jesus Augustin, J.; Buelta, L.; Merino, J.; Merino, R. Bone Morphogenetic Protein and Activin Membrane-Bound Inhibitor, a Transforming Growth Factor beta Rheostat That Controls Murine Treg Cell/Th17 Cell Differentiation and the Development of Autoimmune Arthritis by Reducing Interleukin-2 Signaling. Arthritis Rheumatol. 2016, 68, 1551–1562. [Google Scholar] [CrossRef]
- Sun, S.W.; Chen, L.; Zhou, M.; Wu, J.H.; Meng, Z.J.; Han, H.L.; Miao, S.Y.; Zhu, C.C.; Xiong, X.Z. BAMBI regulates macrophages inducing the differentiation of Treg through the TGF-beta pathway in chronic obstructive pulmonary disease. Respir. Res. 2019, 20, 26. [Google Scholar] [CrossRef]
- Drescher, H.K.; Bartsch, L.M.; Weiskirchen, S.; Weiskirchen, R. Intrahepatic TH17/TReg Cells in Homeostasis and Disease-It’s All About the Balance. Front. Pharmacol. 2020, 11, 588436. [Google Scholar] [CrossRef]
- Liu, C.; Chen, X.; Yang, L.; Kisseleva, T.; Brenner, D.A.; Seki, E. Transcriptional repression of the transforming growth factor beta (TGF-beta) Pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by Nuclear Factor kappaB (NF-kappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J. Biol. Chem. 2014, 289, 7082–7091. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Tao, L.; Xue, D.; Shen, D.; Ma, W.; Zhang, J.; Wang, X.; Zhang, W.; Wu, L.; Pan, K.; Yang, Y.; et al. MicroRNA-942 mediates hepatic stellate cell activation by regulating BAMBI expression in human liver fibrosis. Arch. Toxicol. 2018, 92, 2935–2946. [Google Scholar] [CrossRef]
- Dromann, D.; Rupp, J.; Rohmann, K.; Osbahr, S.; Ulmer, A.J.; Marwitz, S.; Roschmann, K.; Abdullah, M.; Schultz, H.; Vollmer, E.; et al. The TGF-beta.a-pseudoreceptor BAMBI is strongly expressed in COPD lungs and regulated by nontypeable Haemophilus influenzae. Respir. Res. 2010, 11, 67. [Google Scholar] [CrossRef]
- Guillot, N.; Kollins, D.; Gilbert, V.; Xavier, S.; Chen, J.; Gentle, M.; Reddy, A.; Bottinger, E.; Jiang, R.; Rastaldi, M.P.; et al. BAMBI regulates angiogenesis and endothelial homeostasis through modulation of alternative TGFbeta signaling. PLoS ONE 2012, 7, e39406. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Kopec, A.K.; Ray, J.L.; Cline-Fedewa, H.; Nawabi, A.; Schmitt, T.; Nault, R.; Zacharewski, T.R.; Rockwell, C.E.; Flick, M.J.; et al. Fibrin deposition following bile duct injury limits fibrosis through an alphaMbeta2-dependent mechanism. Blood 2016, 127, 2751–2762. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.K.; Abrahams, S.R.; Thornton, S.; Palumbo, J.S.; Mullins, E.S.; Divanovic, S.; Weiler, H.; Owens, A.P., 3rd; Mackman, N.; Goss, A.; et al. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J. Clin. Investig. 2017, 127, 3152–3166. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T. Platelets and fibrin in progression of liver disease: Friends or foes? J. Thromb. Haemost. 2015, 13, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Mitnala, S.; Vishnubhotla, R.K.; Mukherjee, R.; Reddy, D.N.; Rao, P.N. The Riddle of Nonalcoholic Fatty Liver Disease: Progression From Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. J. Clin. Exp. Hepatol. 2015, 5, 147–158. [Google Scholar] [CrossRef]
- Friedman, S.L. A deer in the headlights: BAMBI meets liver fibrosis. Nat. Med. 2007, 13, 1281–1282. [Google Scholar] [CrossRef]
- Chen, M.; Liu, J.; Yang, W.; Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017, 13, 1813–1827. [Google Scholar] [CrossRef]
- Dattaroy, D.; Seth, R.K.; Sarkar, S.; Kimono, D.; Albadrani, M.; Chandrashekaran, V.; Hasson, F.A.; Singh, U.P.; Fan, D.; Nagarkatti, M.; et al. Sparstolonin B (SsnB) attenuates liver fibrosis via a parallel conjugate pathway involving P53-P21 axis, TGF-beta signaling and focal adhesion that is TLR4 dependent. Eur. J. Pharmacol. 2018, 841, 33–48. [Google Scholar] [CrossRef]
- Subramaniam, N.; Sherman, M.H.; Rao, R.; Wilson, C.; Coulter, S.; Atkins, A.R.; Evans, R.M.; Liddle, C.; Downes, M. Metformin-mediated Bambi expression in hepatic stellate cells induces prosurvival Wnt/beta-catenin signaling. Cancer Prev. Res. 2012, 5, 553–561. [Google Scholar] [CrossRef]
- Xavier, S.; Gilbert, V.; Rastaldi, M.P.; Krick, S.; Kollins, D.; Reddy, A.; Bottinger, E.; Cohen, C.D.; Schlondorff, D. BAMBI is expressed in endothelial cells and is regulated by lysosomal/autolysosomal degradation. PLoS ONE 2010, 5, e12995. [Google Scholar] [CrossRef]
- Xie, Y.; Du, D.; Zhang, L.; Yang, Y.; Zou, Z.; Li, Z.; Zhou, L.; Shang, R.; Zhou, P. TJ-M2010-5, A self-developed MyD88 inhibitor, attenuates liver fibrosis by inhibiting the NF-kappaB pathway. Chem. Biol. Interact. 2022, 354, 109839. [Google Scholar] [CrossRef]
- Ten Hove, M.; Pater, L.; Storm, G.; Weiskirchen, S.; Weiskirchen, R.; Lammers, T.; Bansal, R. The hepatic lipidome: From basic science to clinical translation. Adv. Drug Deliv. Rev. 2020, 159, 180–197. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, S.; Zhang, P. Effect of Exogenous Fetuin-A on TGF-beta/Smad Signaling in Hepatic Stellate Cells. Biomed. Res. Int. 2016, 2016, 8462615. [Google Scholar] [CrossRef]
- Sato, M.; Kamada, Y.; Takeda, Y.; Kida, S.; Ohara, Y.; Fujii, H.; Akita, M.; Mizutani, K.; Yoshida, Y.; Yamada, M.; et al. Fetuin-A negatively correlates with liver and vascular fibrosis in nonalcoholic fatty liver disease subjects. Liver Int. 2015, 35, 925–935. [Google Scholar] [CrossRef]
- Dogru, T.; Kirik, A.; Gurel, H.; Rizvi, A.A.; Rizzo, M.; Sonmez, A. The Evolving Role of Fetuin-A in Nonalcoholic Fatty Liver Disease: An Overview from Liver to the Heart. Int. J. Mol. Sci. 2021, 22, 6627. [Google Scholar] [CrossRef]
- Zhao, H.W.; Zhang, Z.F.; Chai, X.; Li, G.Q.; Cui, H.R.; Wang, H.B.; Meng, Y.K.; Liu, H.M.; Wang, J.B.; Li, R.S.; et al. Oxymatrine attenuates CCl4-induced hepatic fibrosis via modulation of TLR4-dependent inflammatory and TGF-beta1 signaling pathways. Int. Immunopharmacol. 2016, 36, 249–255. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Takasaka, N.; Araya, J.; Kurita, Y.; Kobayashi, K.; Ito, S.; Wakui, H.; Yoshii, Y.; Minagawa, S.; Kojima, J.; Hara, H.; et al. Metformin inhibits TGF-b-induced myofibroblast differentiation through AMPK activation. Eur. Respir. J. 2014, 44, P3854. [Google Scholar]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savvidou, S.; Hytiroglou, P.; Orfanou-Koumerkeridou, H.; Panderis, A.; Frantzoulis, P.; Goulis, J. Low serum adiponectin levels are predictive of advanced hepatic fibrosis in patients with NAFLD. J. Clin. Gastroenterol. 2009, 43, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, M.; Lam, K.S.; Xu, A. Protective roles of adiponectin in obesity-related fatty liver diseases: Mechanisms and therapeutic implications. Arq. Bras. Endocrinol. Metabol. 2009, 53, 201–212. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Galle, P.R. Animal models of non-alcoholic steatohepatitis: Of mice and man. Dig. Dis. 2010, 28, 247–254. [Google Scholar] [CrossRef]
- Desai, M.S.; Mariscalco, M.M.; Tawil, A.; Vallejo, J.G.; Smith, C.W. Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J. Leukoc. Biol. 2008, 83, 1336–1344. [Google Scholar] [CrossRef]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef]
- Shockley, K.R.; Witmer, D.; Burgess-Herbert, S.L.; Paigen, B.; Churchill, G.A. Effects of atherogenic diet on hepatic gene expression across mouse strains. Physiol. Genom. 2009, 39, 172–182. [Google Scholar] [CrossRef]
- Velayudham, A.; Dolganiuc, A.; Ellis, M.; Petrasek, J.; Kodys, K.; Mandrekar, P.; Szabo, G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009, 49, 989–997. [Google Scholar] [CrossRef]
- Wooten, J.S.; Poole, K.E.; Harris, M.P.; Guilford, B.L.; Schaller, M.L.; Umbaugh, D.; Seija, A. The effects of voluntary wheel running during weight-loss on biomarkers of hepatic lipid metabolism and inflammation in C57Bl/6J mice. Curr. Res. Physiol. 2022, 5, 63–72. [Google Scholar] [CrossRef]
- Du, J.; Niu, X.; Wang, R.; Zhao, S.; Kong, L.; Zhang, Y.; Nan, Y. TLR4dependent signaling pathway modulation: A novel mechanism by which pioglitazone protects against nutritional fibrotic steatohepatitis in mice. Mol. Med. Rep. 2016, 13, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Oh, I.S.; Tsuchiya, T.; Ohtani, K.I.; Okada, S.; Mori, M. Pioglitazone increases circulating adiponectin levels and subsequently reduces TNF-alpha levels in Type 2 diabetic patients: A randomized study. Diabet. Med. 2006, 23, 253–257. [Google Scholar] [CrossRef]
- Krishnasamy, Y.; Ramshesh, V.K.; Gooz, M.; Schnellmann, R.G.; Lemasters, J.J.; Zhong, Z. Ethanol and High Cholesterol Diet Causes Severe Steatohepatitis and Early Liver Fibrosis in Mice. PLoS ONE 2016, 11, e0163342. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef]
- Schuler-Toprak, S.; Ortmann, O.; Buechler, C.; Treeck, O. The Complex Roles of Adipokines in Polycystic Ovary Syndrome and Endometriosis. Biomedicines 2022, 10, 2503. [Google Scholar] [CrossRef]
- Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. [Google Scholar] [CrossRef]
- Kamada, Y.; Takehara, T.; Hayashi, N. Adipocytokines and liver disease. J. Gastroenterol. 2008, 43, 811–822. [Google Scholar] [CrossRef]
- Rabe, K.; Lehrke, M.; Parhofer, K.G.; Broedl, U.C. Adipokines and insulin resistance. Mol. Med. 2008, 14, 741–751. [Google Scholar] [CrossRef]
- Gupta, A.; Das, A.; Majumder, K.; Arora, N.; Mayo, H.G.; Singh, P.P.; Beg, M.S.; Singh, S. Obesity is Independently Associated With Increased Risk of Hepatocellular Cancer-related Mortality: A Systematic Review and Meta-Analysis. Am. J. Clin. Oncol. 2018, 41, 874–881. [Google Scholar] [CrossRef]
- Luo, X.; Hutley, L.J.; Webster, J.A.; Kim, Y.H.; Liu, D.F.; Newell, F.S.; Widberg, C.H.; Bachmann, A.; Turner, N.; Schmitz-Peiffer, C.; et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI) as a potent negative regulator of adipogenesis and modulator of autocrine/paracrine adipogenic factors. Diabetes 2012, 61, 124–136. [Google Scholar] [CrossRef]
- Choy, L.; Skillington, J.; Derynck, R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 2000, 149, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.R.; Lane, M.D. A role for bone morphogenetic protein-4 in adipocyte development. Cell Cycle 2007, 6, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, C.; Xu, Y.; Huang, K.; Wang, Y.; Wang, X.; Zhou, X.; Pang, W.; Yang, G.; Yu, T. Adipose-specific BMP and activin membrane-bound inhibitor (BAMBI) deletion promotes adipogenesis by accelerating ROS production. J. Biol. Chem. 2021, 296, 100037. [Google Scholar] [CrossRef] [PubMed]
- Duspara, K.; Bojanic, K.; Pejic, J.I.; Kuna, L.; Kolaric, T.O.; Nincevic, V.; Smolic, R.; Vcev, A.; Glasnovic, M.; Curcic, I.B.; et al. Targeting the Wnt Signaling Pathway in Liver Fibrosis for Drug Options: An Update. J. Clin. Transl. Hepatol. 2021, 9, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lu, P.; Ma, Q.; Cao, Y.; Chen, N.; Li, W.; Zhao, S.; Chen, B.; Shi, J.; Sun, Y.; et al. CTNNB1/beta-catenin dysfunction contributes to adiposity by regulating the cross-talk of mature adipocytes and preadipocytes. Sci. Adv. 2020, 6, eaax9605. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T.; Gosens, R. Revisiting asthma therapeutics: Focus on WNT signal transduction. Drug Discov. Today 2018, 23, 49–62. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal. Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Sekiya, T.; Adachi, S.; Kohu, K.; Yamada, T.; Higuchi, O.; Furukawa, Y.; Nakamura, Y.; Nakamura, T.; Tashiro, K.; Kuhara, S.; et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of transforming growth factor-beta signaling, as a target of the beta-catenin pathway in colorectal tumor cells. J. Biol. Chem. 2004, 279, 6840–6846. [Google Scholar] [CrossRef]
- Lin, Z.; Gao, C.; Ning, Y.; He, X.; Wu, W.; Chen, Y.G. The pseudoreceptor BMP and activin membrane-bound inhibitor positively modulates Wnt/beta-catenin signaling. J. Biol. Chem. 2008, 283, 33053–33058. [Google Scholar] [CrossRef]
- Mannaerts, I.; Thoen, L.F.R.; Eysackers, N.; Cubero, F.J.; Batista Leite, S.; Coldham, I.; Colle, I.; Trautwein, C.; van Grunsven, L.A. Unfolded protein response is an early, non-critical event during hepatic stellate cell activation. Cell Death Dis. 2019, 10, 98. [Google Scholar] [CrossRef]
- Mai, Y.; Zhang, Z.; Yang, H.; Dong, P.; Chu, G.; Yang, G.; Sun, S. BMP and activin membrane-bound inhibitor (BAMBI) inhibits the adipogenesis of porcine preadipocytes through Wnt.t/beta-catenin signaling pathway. Biochem. Cell Biol. 2014, 92, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Deldar Abad Paskeh, M.; Mirzaei, S.; Ashrafizadeh, M.; Zarrabi, A.; Sethi, G. Wnt/beta-Catenin Signaling as a Driver of Hepatocellular Carcinoma Progression: An Emphasis on Molecular Pathways. J. Hepatocell. Carcinoma 2021, 8, 1415–1444. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, F.; Catalano, T.; Cotellese, R.; Aceto, G.M. Targeting Wnt/beta-Catenin Pathways in Primary Liver Tumours: From Microenvironment Signaling to Therapeutic Agents. Cancers 2022, 14, 1912. [Google Scholar] [CrossRef] [PubMed]
- Kaposi-Novak, P.; Lee, J.S.; Gomez-Quiroz, L.; Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Investig. 2006, 116, 1582–1595. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Foti, M. PTEN at the crossroad of metabolic diseases and cancer in the liver. Ann. Hepatol. 2008, 7, 192–199. [Google Scholar] [CrossRef]
- Stauffer, J.K.; Scarzello, A.J.; Andersen, J.B.; De Kluyver, R.L.; Back, T.C.; Weiss, J.M.; Thorgeirsson, S.S.; Wiltrout, R.H. Coactivation of AKT and beta-catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res. 2011, 71, 2718–2727. [Google Scholar] [CrossRef]
- Lee, S.; Lee, M.J.; Zhang, J.; Yu, G.R.; Kim, D.G. C-terminal-truncated HBV X promotes hepato-oncogenesis through inhibition of tumor-suppressive beta-catenin/BAMBI signaling. Exp. Mol. Med. 2016, 48, e275. [Google Scholar] [CrossRef]
- Yi, J.; Fan, Y.; Zhang, L.; Wang, H.; Mu, T.; Xie, H.; Gao, H.; Liu, M.; Li, S.; Tang, H. MiR-HCC2 Up-regulates BAMBI and ELMO1 Expression to Facilitate the Proliferation and EMT of Hepatocellular Carcinoma Cells. J. Cancer 2019, 10, 3407–3419. [Google Scholar] [CrossRef]
- Proteinatlas The Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 19 December 2019).
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hu, M.J.; Zhong, X.L.; Ji, L.H.; Wang, J.; Zhang, C.F.; Zhang, R.; Lin, H.M. Screening of a novel autophagy-related prognostic signature and therapeutic targets in hepatocellular carcinoma. J. Gastrointest. Oncol. 2021, 12, 2985–2998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhu, Y. The EpCAM overexpression is associated with clinicopathological significance and prognosis in hepatocellular carcinoma patients: A systematic review and meta-analysis. Int. J. Surg. 2018, 56, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fang, D.; He, Y.; Wei, J. Correlation analysis of tumor mutation burden of hepatocellular carcinoma based on data mining. J. Gastrointest. Oncol. 2021, 12, 1117–1131. [Google Scholar] [CrossRef]
- Studach, L.L.; Menne, S.; Cairo, S.; Buendia, M.A.; Hullinger, R.L.; Lefrancois, L.; Merle, P.; Andrisani, O.M. Subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with HBV X protein. Hepatology 2012, 56, 1240–1251. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Shangguan, L.; Ti, X.; Krause, U.; Hai, B.; Zhao, Y.; Yang, Z.; Liu, F. Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 2012, 30, 2810–2819. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Pioronska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wolfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, M.A.; Chapresto-Garzon, R.; Cadenas, M.; Navarro-Villaran, E.; Negrete, M.; Gomez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntane, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-beta Signalling. J. Signal. Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef] [Green Version]
- Sas, Z.; Cendrowicz, E.; Weinhauser, I.; Rygiel, T.P. Tumor Microenvironment of Hepatocellular Carcinoma: Challenges and Opportunities for New Treatment Options. Int. J. Mol. Sci. 2022, 23, 3778. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2015, 67, 103–117. [Google Scholar] [CrossRef]
- Schutte, K.; Schulz, C.; Poranzke, J.; Antweiler, K.; Bornschein, J.; Bretschneider, T.; Arend, J.; Ricke, J.; Malfertheiner, P. Characterization and prognosis of patients with hepatocellular carcinoma (HCC) in the non-cirrhotic liver. BMC Gastroenterol. 2014, 14, 117. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, F.; Treeck, O.; Mester, P.; Buechler, C. Expression and Function of BMP and Activin Membrane-Bound Inhibitor (BAMBI) in Chronic Liver Diseases and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3473. https://doi.org/10.3390/ijms24043473
Weber F, Treeck O, Mester P, Buechler C. Expression and Function of BMP and Activin Membrane-Bound Inhibitor (BAMBI) in Chronic Liver Diseases and Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(4):3473. https://doi.org/10.3390/ijms24043473
Chicago/Turabian StyleWeber, Florian, Oliver Treeck, Patricia Mester, and Christa Buechler. 2023. "Expression and Function of BMP and Activin Membrane-Bound Inhibitor (BAMBI) in Chronic Liver Diseases and Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 4: 3473. https://doi.org/10.3390/ijms24043473