
Liver Fibrosis Resolution: From Molecular Mechanisms to Therapeutic Opportunities

Abstract
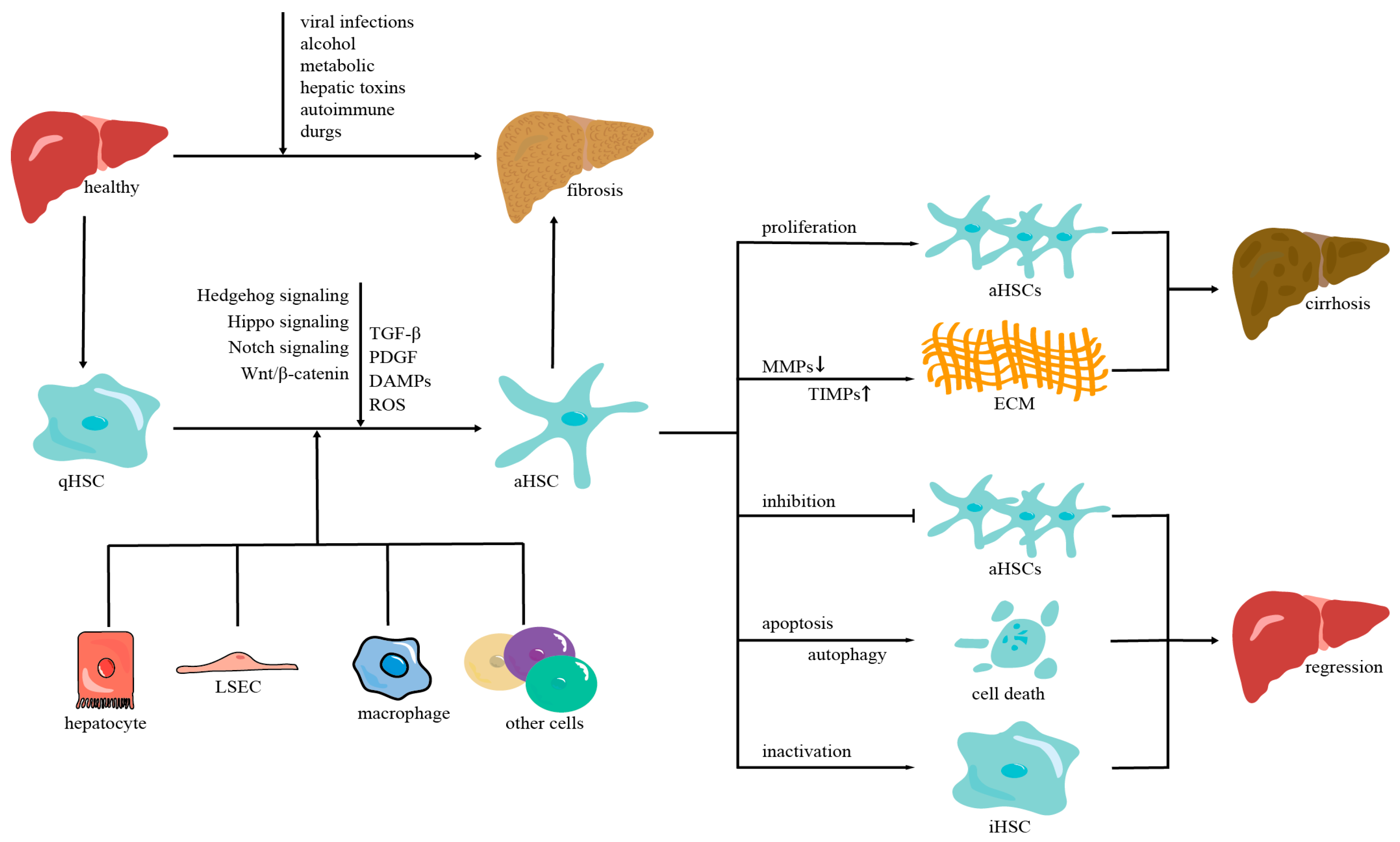
:1. Introduction
2. The Mechanisms of Liver Fibrosis
2.1. Hepatic Stellate Cells
2.2. The Intercellular Crosstalk of HSC Activation
2.2.1. Hepatocytes
2.2.2. Liver Sinusoidal Endothelial Cells
2.2.3. Inflammatory Cells
2.3. The Molecular Mechanisms of HSC Activation
2.3.1. TGF-β Signaling Pathway
2.3.2. PDGF Signaling Pathway
2.3.3. Hippo Signaling Pathway
2.3.4. Reactive Oxygen Species
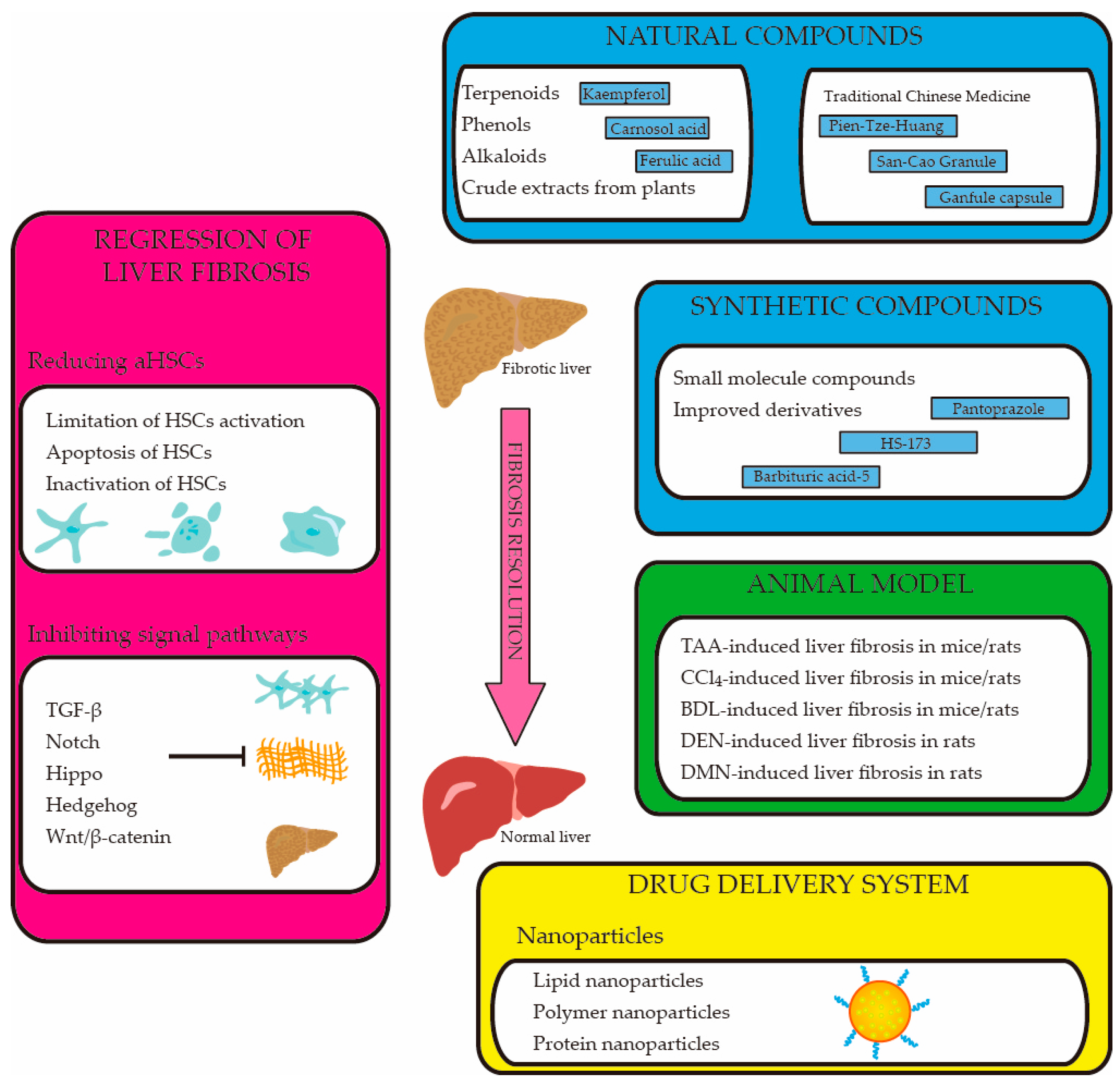
3. Regression of Liver Fibrosis
3.1. Reducing the Number of Activated HSCs
3.1.1. Limitation of HSC Activation
3.1.2. Apoptosis of HSCs
3.1.3. Inactivation of HSCs
3.2. Inhibiting the Liver Fibrosis-Related Signal Pathways
4. The Therapeutic Compounds of Liver Fibrosis
4.1. Natural Compounds

{kind=link}
{kind=link}
| Compounds | Animal Model | Targets/Pathways/Mechanisms | Reference |
|---|---|---|---|
| 18beta-glycyrrhetinic acid (18β-GA) | BDL-induced liver fibrosis in mice | Targeting PRDX1/2 | [19] |
| Resulting in the accumulation of cellular ROS | |||
| Inducing apoptosis in activated HSCs | |||
| Kaempferol (KA) | CCl4-induced liver fibrosis in female C57BL/6 mice | Inhibiting Notch pathway via miR-26b-5p/Jag1 | [116,117,118] |
| Downregulation of TGF-β1/Smad2/3 signaling | |||
| Inhibiting HSC activation | |||
| Swertia purpurascens Wall extract (SPE) | CCl4-induced liver fibrosis in male Wistar rats | Inhibition of TGF-β/Smad/NF-κB signaling | [9] |
| Inhibiting oxidative stress and inflammation | |||
| Demethylzeylasteral (T-96) | CCl4-induced liver fibrosis in male C57BL/6J mice | Suppressing AGAP2-mediated FAK/Akt signaling | [121] |
| Inhibition of downstream TGF-β1 induced fibrosis-related gene expression | |||
| Ferulic acid (FA) | CCl4-induced fibrotic models in male C57BL/6J mice | Inhibiting hepatic oxidative stress, macrophage activation, and HSC activation | [25,120] |
| CCl4-induced liver fibrosis in male Wistar rats | Through PTP1B and AMPK signaling pathways. | ||
| Inhibition of the TGF-β1/Smad pathway | |||
| Methoxyeugenol | CCl4-induced liver fibrosis in male BALB/c mice | Modulating the activated phenotype of HSCs by activating PPAR-γ | [57] |
| A protective effect against oxidative stress damage in hepatocytes | |||
| Benzoquinone derivatives | TAA-induced liver fibrosis in male Balb/C mice | Ameliorating oxidative stress and inflammation | [68] |
| Inducing apoptosis in activated LX-2 cells | |||
| Attenuating liver fibrosis in TAA-induced mice | |||
| Carnosol (CS) | CCl4-induced liver fibrosis in male SD rats | Activation of SIRT1/EZH2 | [28] |
| Inhibiting HSC activation and reversing EMT | |||
| Carnosic acid (CA) | BDL-induced liver fibrosis in male SD rats | Modulating the miR-29b-3p/HMGB1/TLR4 signaling pathway | [7] |
| Attenuating BDL-induced liver fibrosis | |||
| Stachydrine (STA) | CCl4-induced liver fibrosis in male SD rats | Inhibiting inflammation and oxidative stress | [122] |
| Upregulating the ratio of MMPs/TIMPs to promote the degradation of ECM | |||
| Catalpol | CCl4-induced liver fibrosis in male SD rats | Activating autophagy to exert anti-inflammatory effects | [8] |
| Inhibiting the activation of HSCs | |||
| Mogroside IVE (MGIVE) | CCl4-induced liver fibrosis in male C57BL/6 mice | Inhibiting the TLR4 signaling pathway | [112] |
| Reducing inflammatory responses | |||
| Inhibiting TGF-β1 induced HSC activation | |||
| Morin | DEN-induced liver fibrosis in male Wistar rats | Activating HIPPO signaling | [53] |
| Downregulating TGF-β signaling | |||
| Attenuating fibrillar collagen deposition | |||
| Preventing HSC activation | |||
| S-allyl cysteine (SAC) | CCl4-induced liver fibrosis in male Wistar rats | Inhibiting the expression of TGF-β | [123] |
| A decrease in the profibrogenic cytokine, TGF-β | |||
| Correcting cytokine abnormality | |||
| SMND-309 | CCl4-induced liver fibrosis in male SD rats | Suppressing the expression of CTGF | [51] |
| Scavenging lipid peroxidation products | |||
| Increasing endogenous anti-oxidation enzyme activity | |||
| Quercetin (QE) | CCl4-induced liver fibrosis in male C57 mice | Suppressing the TGF-β1/Smads signaling pathway | [124] |
| BDL-induced liver fibrosis in male C57 mice | Activating the PI3K/Akt signaling pathway to inhibit autophagy | ||
| Attenuating HSC activation | |||
| Tormentic acid (TA) | CCl4-induced liver fibrosis in male SD rats | Suppressing HSC activation | [125] |
| Suppressing the PI3K/Akt/mTOR signaling pathway | |||
| Inhibiting the NF-κB signaling pathway | |||
| Gomisin D | CCl4-induced liver fibrosis in male Balb/C mice | Inhibiting HSC proliferation and activation | [82] |
| Promoting HSC apoptosis | |||
| Regulating the PDGF-BB/PDGFRβ signaling pathway | |||
| Costunolide (COS) | CCl4-induced liver fibrosis in male Balb/C mice | Regulating the Notch3–HES1 pathway | [111] |
| BDL-induced liver fibrosis in male SD rats | Disturbing the PPM1G/WWP2 complex | ||
| Blocking the inhibitory effect of PPM1G on WWP2 | |||
| 5-Methoxytryptophan (5-MTP) | CCl4-induced liver fibrosis in male SD rats | Regulation of the FOXO3a/miR-21/ATG5 pathway | [126] |
| Inducing autophagy and suppressing HSCs’ activation | |||
| Physalin B | CCl4-induced liver fibrosis in male C57BL/6J mice | Disrupting LAP2α/HDAC1 complexes | [127] |
| BDL-induced liver fibrosis in male C57BL/6J mice | Inhibiting HDAC1-mediated GLI1 deacetylation | ||
| Inhibiting HSC activation | |||
| Ginsenoside Rg1 (G-Rg1) | CCl4-induced liver fibrosis male C57BL/6J mice | Attenuating IDO1-mediated inhibition of maturation of DCs | [128] |
| Inhibiting the proliferation of HSCs | |||
| Germarcone (GER) | MCS/MCD diet induction in male C57BL/6 mice | Reducing the release of ROS | [129] |
| Regulating TGF-β/Smad and apoptosis pathways | |||
| Inhibiting the activation and survival of HSCs | |||
| Sweroside | CCl4-induced liver fibrosis in male C57BL/6 mice | Regulation of FXR-miR-29a signaling pathway | [130] |
| Inhibition of HSC proliferation | |||
| Artesunate | CCl4-induced liver fibrosis in male ICR mice | Activating HSC ferroptosis | [91] |
| Inhibiting HSC activation | |||
| Linderalactone (LIN) | CCl4-induced liver fibrosis in C57BL/6 mice | Inhibiting TGF-β/Smad signaling | [131] |
| Suppressing HSC activation |
4.2. Synthetic Compounds
4.3. Drug Delivery System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, W.; Cheng, S.; Bao, Z.; Wang, Y.; Zhou, W.; Wang, J.; Yang, Q.; Chen, C.; Wang, W. Advances in the research of nanodrug delivery system for targeted treatment of liver fibrosis. Biomed. Pharmacother. 2021, 137, 111342. [Google Scholar] [CrossRef] [PubMed]
- Hadi, F.; Awan, S.J.; Tayyeb, A.; Maqbool, T.; Shehzadi, S.; Malik, S.; Kausar, H.; Malik, A. Hepato-protective role of itraconazole mediated cytochrome p450 pathway inhibition in liver fibrosis. Pak. J. Pharm. Sci. 2020, 33, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.C.; Bai, J.; Han, H.; Qin, H.Y. The versatility of macrophage heterogeneity in liver fibrosis. Front. Immunol. 2022, 13, 968879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Q.S.; Dahan, A.; Xue, J.Y.; Wei, L.W.; Tan, W.F.; Zhang, G.Q. Transcriptomic analyses reveal the molecular mechanisms of schisandrin B alleviates CCl4-induced liver fibrosis in rats by RNA-sequencing. Chem. Biol. Interact. 2019, 309, 108675. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q.; He, J.; Li, Y. Novel Therapeutic Targets in Liver Fibrosis. Front. Mol. Biosci. 2021, 8, 766855. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z.C.; Zhu, J.; Xu, T.; Zhao, Y.; Zhao, H.Y.; Tang, F.; Li, Z.L.; Zhou, J.J.; Gao, D.Y.; et al. Carnosic Acid Alleviates BDL-Induced Liver Fibrosis through miR-29b-3p-Mediated Inhibition of the High-Mobility Group Box 1/Toll-Like Receptor 4 Signaling Pathway in Rats. Front. Pharmacol. 2018, 8, 976. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhu, P.; Zhang, L.; Xiong, B.; Tao, J.; Guan, W.; Li, C.; Chen, C.; Gu, J.; Duanmu, J.; et al. Autophagy inhibition attenuates the induction of anti-inflammatory effect of catalpol in liver fibrosis. Biomed. Pharmacother. 2018, 103, 1262–1271. [Google Scholar] [CrossRef]
- Raj, D.; Sharma, V.; Upadhyaya, A.; Kumar, N.; Joshi, R.; Acharya, V.; Kumar, D.; Patial, V. Swertia purpurascens Wall ethanolic extract mitigates hepatic fibrosis and restores hepatic hepcidin levels via inhibition of TGF beta/SMAD/NF kappa B signaling in rats. J. Ethnopharmacol. 2022, 284, 114741. [Google Scholar] [CrossRef]
- Milito, A.; Brancaccio, M.; D’Argenio, G.; Castellano, I. Natural Sulfur-Containing Compounds: An Alternative Therapeutic Strategy against Liver Fibrosis. Cells 2019, 8, 1356. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Guo, C.Y.; Wu, J.Y. Astaxanthin in Liver Health and Disease: A Potential Therapeutic Agent. Drug. Des. Dev. Ther. 2020, 14, 2275–2285. [Google Scholar] [CrossRef]
- Kaftanovskaya, E.M.; Ng, H.H.; Soula, M.; Rivas, B.; Myhr, C.; Ho, B.A.; Cervantes, B.A.; Shupe, T.D.; Devarasetty, M.; Hu, X.; et al. Therapeutic effects of a small molecule agonist of the relaxin receptor ML290 in liver fibrosis. Faseb J. 2019, 33, 12435–12446. [Google Scholar] [CrossRef] [Green Version]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.R.; Roper, J.A.; Grove, J.I.; Aithal, G.P.; Pun, K.T.; Bennett, A.J. Integrins as a drug target in liver fibrosis. Liver Int. 2022, 42, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, F.; Wu, J.; Zhuge, Y. Nanotechnology in Drug Delivery for Liver Fibrosis. Front. Mol. Biosci. 2021, 8, 804396. [Google Scholar] [CrossRef] [PubMed]
- Bestion, E.; Jilkova, Z.M.; Mege, J.L.; Novello, M.; Kurma, K.; Pour, S.T.A.; Lalmanach, G.; Vanderlynden, L.; Fizanne, L.; Bassissi, F.; et al. GNS561 acts as a potent anti-fibrotic and pro-fibrolytic agent in liver fibrosis through TGF-beta 1 inhibition. Ther. Adv. Chronic Dis. 2020, 11, 2040622320942042. [Google Scholar] [CrossRef]
- Feng, M.; Ding, J.; Wang, M.; Zhang, J.; Zhu, X.; Guan, W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int. J. Biol. Sci. 2018, 14, 1033–1040. [Google Scholar] [CrossRef]
- Itaba, N.; Kono, Y.; Watanabe, K.; Yokobata, T.; Oka, H.; Osaki, M.; Kakuta, H.; Morimoto, M.; Shiota, G. Reversal of established liver fibrosis by IC-2-engineered mesenchymal stem cell sheets. Sci. Rep. 2019, 9, 6841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Luo, P.; Zheng, L.H.; Chen, J.Y.; Zhang, J.Z.; Tang, H.; Liu, D.D.; He, X.L.; Shi, Q.L.; Gu, L.W.; et al. 18beta-glycyrrhetinic acid induces ROS-mediated apoptosis to ameliorate hepatic fibrosis by targeting PRDX1/2 in activated HSCs. J. Pharm. Anal. 2022, 12, 570–582. [Google Scholar] [CrossRef]
- Li, W.Y.; Chen, J.Y.; Sun, C.; Sparks, R.P.; Pantano, L.; Rahman, R.U.; Moran, S.P.; Pondick, J.V.; Kirchner, R.; Wrobel, D.; et al. Nanchangmycin regulates FYN, PTK2, and MAPK1/3 to control the fibrotic activity of human hepatic stellate cells. eLife 2022, 11, e74513. [Google Scholar] [CrossRef]
- Wu, K.K. Control of Tissue Fibrosis by 5-Methoxytryptophan, an Innate Anti-Inflammatory Metabolite. Front. Pharmacol. 2021, 12, 759199. [Google Scholar] [CrossRef]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef]
- Wang, F.D.; Zhou, J.; Chen, E.Q. Molecular Mechanisms and Potential New Therapeutic Drugs for Liver Fibrosis. Front. Pharmacol. 2022, 13, 787748. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, X.; Wang, Z.; Yin, M.; Zhao, Z.; Li, Y.; Li, W. Pokeweed antiviral protein attenuates liver fibrosis in mice through regulating Wnt/Jnk mediated glucose metabolism. Saudi J. Gastroenterol. 2018, 24, 157–164. [Google Scholar] [CrossRef]
- Wu, J.Z.; Xue, X.Y.; Fan, G.F.; Gu, Y.Q.; Zhou, F.; Zheng, Q.; Liu, R.P.; Li, Y.J.; Ma, B.N.; Li, S.; et al. Ferulic Acid Ameliorates Hepatic Inflammation and Fibrotic Liver Injury by Inhibiting PTP1B Activity and Subsequent Promoting AMPK Phosphorylation. Front. Pharmacol. 2021, 12, 754976. [Google Scholar] [CrossRef]
- Karimi, J.; Mohammadalipour, A.; Sheikh, N.; Khodadadi, I.; Hashemnia, M.; Goudarzi, F.; Khanjarsim, V.; Solgi, G.; Hajilooi, M.; Bahabadi, M.; et al. Protective effects of combined Losartan and Nilotinib on carbon tetrachloride (CCl4)-induced liver fibrosis in rats. Drug. Chem. Toxicol. 2020, 43, 468–478. [Google Scholar] [CrossRef]
- Ding, N.; Wei, B.; Fu, X.H.; Wang, C.; Wu, Y.M. Natural Products that Target the NLRP3 Inflammasome to Treat Fibrosis. Front. Pharmacol. 2020, 11, 591393. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Wang, Z.C.; Tang, F.; Zhao, Y.; Feng, D.C.; Li, Y.; Hu, Y.; Wang, C.; Zhou, J.J.; Tian, X.F.; et al. Carnosol-mediated Sirtuin 1 activation inhibits Enhancer of Zeste Homolog 2 to attenuate liver fibrosis. Pharmacol. Res. 2018, 128, 327–337. [Google Scholar] [CrossRef]
- Wei, M.J.; Zhang, Y.; Zhang, H.; Huang, Z.L.; Miao, H.; Zhang, T.Y.; Lu, B.; Ji, L.L. HMGB1 induced endothelial to mesenchymal transition in liver fibrosis: The key regulation of early growth response factor 1. Biochim. Et Biophys. Acta-Gen. Subj. 2022, 1866, 130202. [Google Scholar] [CrossRef]
- Garbuzenko, D.V. Pathophysiological mechanisms of hepatic stellate cells activation in liver fibrosis. World J. Clin. Cases 2022, 10, 3662–3676. [Google Scholar] [CrossRef]
- Nokkeaw, A.; Thamjamrassri, P.; Tangkijvanich, P.; Ariyachet, C. Regulatory Functions and Mechanisms of Circular RNAs in Hepatic Stellate Cell Activation and Liver Fibrosis. Cells 2023, 12, 378. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ge, Q.; Zhang, Y.; Li, B.; Cheng, E.; Wang, Y.; Huang, Y. A review of the effect of exosomes from different cells on liver fibrosis. Biomed. Pharmacother. 2023, 161, 114415. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Quan, J. Exosomes derived from natural killer cells inhibit hepatic stellate cell activation and liver fibrosis. Hum. Cell 2020, 33, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, Y.; Yang, Y.; Liu, K.; Wu, J.; Gao, P.; Zhang, C. Exosomes in liver fibrosis: The role of modulating hepatic stellate cells and immune cells, and prospects for clinical applications. Front. Immunol. 2023, 14, 1133297. [Google Scholar] [CrossRef] [PubMed]
- Pi, L.; Sun, C.; Jn-Simon, N.; Basha, S.; Thomas, H.; Figueroa, V.; Zarrinpar, A.; Cao, Q.; Petersen, B. CCN2/CTGF promotes liver fibrosis through crosstalk with the Slit2/Robo signaling. J. Cell. Commun. Signal. 2023, 17, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qi, Y.F.; Yu, Y.R. STAT3: A key regulator in liver fibrosis. Ann. Hepatol. 2021, 21, 100224. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell. Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Ni, Y.A.; Chen, H.; Nie, H.; Zheng, B.; Gong, Q. HMGB1: An overview of its roles in the pathogenesis of liver disease. J. Leukoc. Biol. 2021, 110, 987–998. [Google Scholar] [CrossRef]
- Li, J.; Zeng, C.; Zheng, B.; Liu, C.; Tang, M.; Jiang, Y.; Chang, Y.; Song, W.; Wang, Y.; Yang, C. HMGB1-induced autophagy facilitates hepatic stellate cells activation: A new pathway in liver fibrosis. Clin. Sci. 2018, 132, 1645–1667. [Google Scholar] [CrossRef]
- Luo, X.; Xu, Z.X.; Wu, J.C.; Luo, S.Z.; Xu, M.Y. Hepatocyte-derived exosomal miR-27a activateshepatic stellate cells through the inhibitionof PINK1-mediated mitophagy in MAFLD. Mol. Ther. Nucleic Acids 2021, 26, 1241–1254. [Google Scholar] [CrossRef]
- Ding, B.S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927.e916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 173–179. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Abdelghffar, E.A.; Obaid, W.A.; Alamoudi, M.O.; Mohammedsaleh, Z.M.; Annaz, H.; Abdelfattah, M.A.O.; Sobeh, M. Thymus fontanesii attenuates CCl4-induced oxidative stress and inflammation in mild liver fibrosis. Biomed. Pharmacother. 2022, 148, 112738. [Google Scholar] [CrossRef]
- Liu, C.; Tao, Q.; Sun, M.; Wu, J.Z.; Yang, W.; Jian, P.; Peng, J.; Hu, Y.; Liu, C.; Liu, P. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab. Invest. 2010, 90, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Tian, J.; Jiang, W.; Gao, Y.; Fu, F. Therapeutic effects of SMND-309, a new metabolite of salvianolic acid B, on experimental liver fibrosis. Eur. J. Pharmacol. 2011, 650, 390–395. [Google Scholar] [CrossRef]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-beta1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Perumal, N.; Perumal, M.; Halagowder, D.; Sivasithamparam, N. Morin attenuates diethylnitrosamine-induced rat liver fibrosis and hepatic stellate cell activation by co-ordinated regulation of Hippo/Yap and TGF-beta1/Smad signaling. Biochimie 2017, 140, 10–19. [Google Scholar] [CrossRef]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-beta signaling in fibrosis: Mechanisms and targets. Semin. Cell. Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Zhang, J.H.; Li, Y.P.; Liu, Q.H.; Huang, Y.; Li, R.; Wu, T.; Zhang, Z.J.; Zhou, J.; Huang, H.; Tang, Q.; et al. Sirt6 Alleviated Liver Fibrosis by Deacetylating Conserved Lysine 54 on Smad2 in Hepatic Stellate Cells. Hepatology 2021, 73, 1140–1157. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor beta-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef] [Green Version]
- Basso, B.D.; Haute, G.V.; Ortega-Ribera, M.; Luft, C.; Antunes, G.L.; Bastos, M.S.; Carlessi, L.P.; Levorse, V.G.; Cassel, E.; Donadio, M.V.F.; et al. Methoxyeugenol deactivates hepatic stellate cells and attenuates liver fibrosis and inflammation through a PPAR-gamma and NF-kB mechanism. J. Ethnopharmacol. 2021, 280, 114433. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Zhang, Z.; Mu, Y.; Zhang, J.; Zhou, Y.; Cattaneo, P.; Veevers, J.; Peter, A.K.; Manso, A.M.; Knowlton, K.U.; Zhou, X.; et al. Kindlin-2 Is Essential for Preserving Integrity of the Developing Heart and Preventing Ventricular Rupture. Circulation 2019, 139, 1554–1556. [Google Scholar] [CrossRef]
- Yu, J.; Hu, Y.; Gao, Y.; Li, Q.; Zeng, Z.; Li, Y.; Chen, H. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell. Death Discov. 2018, 4, 34. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of Action and In Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Riehle, K.J.; Shimizu-Albergine, M.; Bauer, R.L.; Hudkins, K.L.; Johansson, F.; Yeh, M.M.; Mahoney, W.M., Jr.; Yeung, R.S.; Campbell, J.S. Activation of platelet-derived growth factor receptor alpha contributes to liver fibrosis. PLoS ONE 2014, 9, e92925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Russell, J.O.; Camargo, F.D. Hippo signalling in the liver: Role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 297–312. [Google Scholar] [CrossRef]
- Acharya, P.; Chouhan, K.; Weiskirchen, S.; Weiskirchen, R. Cellular Mechanisms of Liver Fibrosis. Front. Pharmacol. 2021, 12, 671640. [Google Scholar] [CrossRef]
- Miao, Y.; Wu, Y.L.; Jin, Y.J.; Lei, M.Z.; Nan, J.X.; Wu, X. Benzoquinone derivatives with antioxidant activity inhibit activated hepatic stellate cells and attenuate liver fibrosis in TAA-induced mice. Chem.-Biol. Interact. 2020, 317, 108945. [Google Scholar] [CrossRef]
- Zheng, M.; Li, Y.Y.; Wang, G.F.; Jin, J.Y.; Wang, Y.H.; Wang, T.M.; Yang, L.; Liu, S.Y.; Wu, J.S.; Wang, Z.T.; et al. Protective effect of cultured bear bile powder against dimethylnitrosamine-induced hepatic fibrosis in rats. Biomed. Pharmacother. 2019, 112, 108701. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Issa, R.; Williams, E.; Trim, N.; Kendall, T.; Arthur, M.J.P.; Reichen, J.; Benyon, R.C.; Iredale, J.P. Apoptosis of hepatic stellate cells: Involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 2001, 48, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair. 2015, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.W.; Liu, H.L.; Ke, B.L.; Jiang, J.; Wu, B. The peripheral CB1 receptor antagonist JD5037 attenuates liver fibrosis via a CB1 receptor/beta-arrestin1/Akt pathway. Br. J. Pharmacol. 2020, 177, 2830–2847. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Hua, L.P.; Lin, C.F.; Yuan, M.Z.; Xu, W.; Raj, A.D.; Venkidasamy, B.; Cespedes-Acuna, C.L.; Nile, S.H.; Yan, G.H.; et al. Pien-Tze-Huang alleviates CCl4-induced liver fibrosis through the inhibition of HSC autophagy and the TGF-beta 1/Smad2 pathway. Front. Pharmacol. 2022, 13, 937484. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, F.; Chen, P.; Li, S.; Gu, Y.; Wang, L.; Chen, C.; Yuan, Y. Gomisin D alleviates liver fibrosis through targeting PDGFRbeta in hepatic stellate cells. Int. J. Biol. Macromol. 2023, 235, 123639. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, N.; Villamayor, L.; Diaz, I.; Carmona, R.; Ramos-Rodriguez, M.; Munoz-Chapuli, R.; Pasquali, L.; Toscano, M.G.; Martin, F.; Cano, D.A.; et al. GATA4 induces liver fibrosis regression by deactivating hepatic stellate cells. JCI Insight 2021, 6, e150059. [Google Scholar] [CrossRef]
- Jelski, W.; Szmitkowski, M. Alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) in the cancer diseases. Clin. Chim. Acta 2008, 395, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Li, J.; Li, M.R.; Qi, B.; Wang, Z.; Wang, G.J.; Gao, J.; Qiao, H.L. Higher Activity of Alcohol Dehydrogenase Is Correlated with Hepatic Fibrogenesis. J. Pharmacol. Exp. Ther. 2018, 367, 473–482. [Google Scholar] [CrossRef]
- Zhao, T.; Han, Z.; Zhang, J.; Ding, Y.; Chen, J.; Qiao, H.; Gao, N. Effect of ADHI on hepatic stellate cell activation and liver fibrosis in mice. Biochem. Biophys. Res. Commun. 2023, 651, 98–106. [Google Scholar] [CrossRef]
- Wu, B.; Huang, L.; Wang, Y.; Zeng, L.; Lin, Y.; Li, J.; Wang, S.; Zhang, G.; An, L. Yao medicine Amydrium hainanense suppresses hepatic fibrosis by repressing hepatic stellate cell activation via STAT3 signaling. Front. Pharmacol. 2022, 13, 1043022. [Google Scholar] [CrossRef]
- Meng, H.; Jiang, L.; Jia, P.; Niu, R.; Bu, F.; Zhu, Y.; Pan, X.; Li, J.; Liu, J.; Zhang, Y.; et al. Inhibition of circular RNA ASPH reduces the proliferation and promotes the apoptosis of hepatic stellate cells in hepatic fibrosis. Biochem. Pharmacol. 2023, 210, 115451. [Google Scholar] [CrossRef]
- Ma, L.; Wei, J.F.; Zeng, Y.L.; Liu, J.P.; Xiao, E.H.; Kang, Y.H.; Kang, Y. Mesenchymal stem cell-originated exosomal circDIDO1 suppresses hepatic stellate cell activation by miR-141-3p/PTEN/AKT pathway in human liver fibrosis. Drug. Deliv. 2022, 29, 440–453. [Google Scholar] [CrossRef]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef]
- Gonzalez, S.A.; Fiel, M.I.; Sauk, J.; Canchis, P.W.; Liu, R.C.; Chiriboga, L.; Yee, H.T.; Jacobson, I.M.; Talal, A.H. Inverse association between hepatic stellate cell apoptosis and fibrosis in chronic hepatitis C virus infection. J. Viral Hepat. 2009, 16, 141–148. [Google Scholar] [CrossRef]
- Koda, Y.; Teratani, T.; Chu, P.S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. [Google Scholar] [CrossRef]
- Xu, T.; Ni, M.M.; Xing, L.; Li, X.F.; Meng, X.M.; Huang, C.; Li, J. NLRC5 regulates TGF-beta1-induced proliferation and activation of hepatic stellate cells during hepatic fibrosis. Int. J. Biochem. Cell. Biol. 2016, 70, 92–104. [Google Scholar] [CrossRef]
- Liu, H.; Dai, L.; Wang, M.; Feng, F.; Xiao, Y. Tunicamycin Induces Hepatic Stellate Cell Apoptosis Through Calpain-2/Ca(2+)-Dependent Endoplasmic Reticulum Stress Pathway. Front. Cell. Dev. Biol. 2021, 9, 684857. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, S.M.; Hur, W.; Kang, B.Y.; Lee, H.L.; Nam, H.; Yoo, S.H.; Sung, P.S.; Kwon, J.H.; Jang, J.W.; et al. Tenofovir disoproxil fumarate directly ameliorates liver fibrosis by inducing hepatic stellate cell apoptosis via downregulation of PI3K/Akt/mTOR signaling pathway. PLoS ONE 2021, 16, e0261067. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728–1744.e1714. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Choi, S.; Lim, C.; Lee, H.; Oh, J. Albumin mediates PPAR-gamma or C/EBP-alpha-induced phenotypic changes in pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2010, 391, 640–644. [Google Scholar] [CrossRef]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K.; Tsukamoto, H. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, H.F., Jr.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Kamiya, A.; Sumiyoshi, H.; Tsuruya, K.; Kagawa, T.; Inagaki, Y. A Deactivation Factor of Fibrogenic Hepatic Stellate Cells Induces Regression of Liver Fibrosis in Mice. Hepatology 2020, 71, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Suk, F.M.; Liu, C.L.; Chen, T.L.; Twu, Y.C.; Hsu, M.H.; Liao, Y.J. Antifibrotic Effects of a Barbituric Acid Derivative on Liver Fibrosis by Blocking the NF-kappa B Signaling Pathway in Hepatic Stellate Cells. Front. Pharmacol. 2020, 11, 388. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/beta-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Kruger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813. [Google Scholar] [CrossRef] [Green Version]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, N.; Qiu, T.; Sun, X. The Role of Autophagy and NLRP3 Inflammasome in Liver Fibrosis. Biomed. Res. Int. 2020, 2020, 7269150. [Google Scholar] [CrossRef]
- Song, Y.R.; Jang, M.H.; Jang, B.; Bae, S.J.; Bak, S.B.; Lee, S.M.; Yun, U.-J.; Lee, J.H.; Park, S.M.; Jung, D.H.; et al. Jageum-Jung, the herbal pharmaceuticals, inhibits the hepatic fibrogenesis as mediated with TGF-β1/smad signaling. Mol. Cell. Toxicol. 2022, 18, 243–251. [Google Scholar] [CrossRef]
- Dewidar, B.; Soukupova, J.; Fabregat, I.; Dooley, S. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis: Updated. Curr. Pathobiol. Rep. 2015, 3, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.X.; Liu, H.T.; Zhang, N.; Niu, W.X.; Lu, Z.N.; Bao, Y.Y.; Huang, R.; Yu, D.K.; Shao, R.G.; He, H.W. Costunolide represses hepatic fibrosis through WW domain-containing protein 2-mediated Notch3 degradation. Br. J. Pharmacol. 2020, 177, 372–387. [Google Scholar] [CrossRef]
- Cao, F.Y.; Zhang, Y.F.; Li, W.G.; Shimizu, K.S.; Xie, H.F.; Zhang, C.F. Mogroside IVE attenuates experimental liver fibrosis in mice and inhibits HSC activation through downregulating TLR4-mediated pathways. Int. Immunopharmacol. 2018, 55, 183–192. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, X. Antibacterial activity of 18beta-glycyrrhetinic acid against Neisseria gonorrhoeae in vitro. Biochem. Biophys. Rep. 2023, 33, 101427. [Google Scholar] [CrossRef]
- Kumar, A.; Archo, S.; Singh, C.P.; Naikoo, S.H.; Singh, B.; Kaur, S.; Tasduq, S.A. Photoprotective effect of 18beta-glycyrrhetinic acid derivatives against ultra violet (UV)-B-Induced skin aging. Bioorg Med. Chem. Lett. 2022, 76, 128984. [Google Scholar] [CrossRef]
- Du, Y.C.; Lai, L.; Zhang, H.; Zhong, F.R.; Cheng, H.L.; Qian, B.L.; Tan, P.; Xia, X.M.; Fu, W.G. Kaempferol from Penthorum chinense Pursh suppresses HMGB1/TLR4/NF-kappaB signaling and NLRP3 inflammasome activation in acetaminophen-induced hepatotoxicity. Food Funct. 2020, 11, 7925–7934. [Google Scholar] [CrossRef]
- Xiao, X.L.; Hu, Q.C.; Deng, X.Y.; Shi, K.Y.; Zhang, W.W.; Jiang, Y.X.; Ma, X.; Zeng, J.H.; Wang, X.Y. Old wine in new bottles: Kaempferol is a promising agent for treating the trilogy of liver diseases. Pharmacol. Res. 2022, 175, 106005. [Google Scholar] [CrossRef]
- Xu, T.; Huang, S.; Huang, Q.; Ming, Z.; Wang, M.; Li, R.; Zhao, Y. Kaempferol attenuates liver fibrosis by inhibiting activin receptor-like kinase 5. J. Cell. Mol. Med. 2019, 23, 6403–6410. [Google Scholar] [CrossRef]
- Zhou, G.; Li, C.; Zhang, R.; Zhan, Y.; Lin, L.; Lang, Z.; Tao, Q.; Zheng, J. Kaempferol Inhibits Hepatic Stellate Cell Activation by Regulating miR-26b-5p/Jag1 Axis and Notch Pathway. Front. Pharmacol. 2022, 13, 881855. [Google Scholar] [CrossRef]
- Wei, Z.; Xue, Y.; Xue, Y.; Cheng, J.; Lv, G.; Chu, L.; Ma, Z.; Guan, S. Ferulic acid attenuates non-alcoholic steatohepatitis by reducing oxidative stress and inflammation through inhibition of the ROCK/NF-kappaB signaling pathways. J. Pharmacol. Sci. 2021, 147, 72–80. [Google Scholar] [CrossRef]
- Mu, M.; Zuo, S.; Wu, R.M.; Deng, K.S.; Lu, S.; Zhu, J.J.; Zou, G.L.; Yang, J.; Cheng, M.L.; Zhao, X.K. Ferulic acid attenuates liver fibrosis and hepatic stellate cell activation via inhibition of TGF-beta/Smad signaling pathway. Drug. Des. Dev. Ther. 2018, 12, 4107–4115. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Guo, W.R.; Li, R.X.; Han, Y.Q.; Gao, Q.; Wang, S.Z. Demethylzeylasteral attenuates hepatic stellate cell activation and liver fibrosis by inhibiting AGAP2 mediated signaling. Phytomedicine 2022, 105, 154349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Yang, A.H.; Wu, Y.; Guan, W.; Xiong, B.; Peng, X.Q.; Wei, X.J.; Chen, C.; Liu, Z.G. Stachydrine ameliorates carbon tetrachloride-induced hepatic fibrosis by inhibiting inflammation, oxidative stress and regulating MMPs/TIMPs system in rats. Biomed. Pharmacother. 2018, 97, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Kodai, S.; Takemura, S.; Kubo, S.; Azuma, H.; Minamiyama, Y. Therapeutic administration of an ingredient of aged-garlic extracts, S-allyl cysteine resolves liver fibrosis established by carbon tetrachloride in rats. J. Clin. Biochem. Nutr. 2015, 56, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, Q.; Mo, W.; Feng, J.; Li, S.; Li, J.; Liu, T.; Xu, S.; Wang, W.; Lu, X.; et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci. Rep. 2017, 7, 9289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Wei, Y.; Li, Y.; Xiong, Y.; Fang, B.; Li, C.; Huang, Q.; Huang, R.; Wei, J. Tormentic Acid Ameliorates Hepatic Fibrosis in vivo by Inhibiting Glycerophospholipids Metabolism and PI3K/Akt/mTOR and NF-kappaB Pathways: Based on Transcriptomics and Metabolomics. Front. Pharmacol. 2022, 13, 801982. [Google Scholar] [CrossRef]
- Tong, M.; Zheng, Q.; Liu, M.; Chen, L.; Lin, Y.H.; Tang, S.G.; Zhu, Y.M. 5-methoxytryptophan alleviates liver fibrosis by modulating FOXO3a/miR-21/ATG5 signaling pathway mediated autophagy. Cell. Cycle 2021, 20, 676–688. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Ye, S.T.; Yu, D.K.; Zhang, Y.Q.; Li, J.; Zhang, M.H.; Leng, Y.R.; Yang, T.; Luo, J.G.; Chen, X.L.; et al. Physalin B attenuates liver fibrosis via suppressing LAP2 alpha-HDAC1-mediated deacetylation of the transcription factor GLI1 and hepatic stellate cell activation. Br. J. Pharmacol. 2021, 178, 3428–3447. [Google Scholar] [CrossRef]
- Mo, C.; Xie, S.W.; Zeng, T.; Lai, Y.Q.; Huang, S.; Zhou, C.Y.; Yan, W.X.; Huang, S.H.; Gao, L.; Lv, Z.P. Ginsenoside-Rg1 acts as an IDO1 inhibitor, protects against liver fibrosis via alleviating IDO1-mediated the inhibition of DCs maturation. Phytomedicine 2021, 84, 153524. [Google Scholar] [CrossRef]
- Li, Z.Y.; Wang, Z.L.; Dong, F.; Shi, W.; Dai, W.Z.; Zhao, J.; Li, Q.; Fang, Z.E.; Ren, L.T.; Liu, T.T.; et al. Germacrone Attenuates Hepatic Stellate Cells Activation and Liver Fibrosis via Regulating Multiple Signaling Pathways. Front. Pharmacol. 2021, 12, 745561. [Google Scholar] [CrossRef]
- Gong, J.T.; Yang, F.; Yang, Q.L.; Tang, X.W.; Shu, F.F.; Xu, L.M.; Wang, Z.T.; Yang, L. Sweroside ameliorated carbon tetrachloride (CCl4)-induced liver fibrosis through FXR-miR-29a signaling pathway. J. Nat. Med. 2020, 74, 17–25. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, B.; Li, Y.; Xu, Y.; Wang, Y.; Zheng, L.; Zheng, X.; Yin, L.; Chen, G.; Wang, Y.; et al. Identification of linderalactone as a natural inhibitor of SHP2 to ameliorate CCl(4)-induced liver fibrosis. Front. Pharmacol. 2023, 14, 1098463. [Google Scholar] [CrossRef]
- Wei, S.; Niu, M.; Wang, J.; Wang, J.; Su, H.; Luo, S.; Zhang, X.; Guo, Y.; Liu, L.; Liu, F.; et al. A network pharmacology approach to discover active compounds and action mechanisms of San-Cao Granule for treatment of liver fibrosis. Drug. Des. Devel Ther. 2016, 10, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.Q.; Pan, Y.F.; Yang, J.Y.; Li, R.S.; Yuan, C.L.; Liao, Y.F.; Zhang, D.D.; Liu, C.H. Antiliver Fibrosis Formula of Fuzheng Huayu Alleviates Inflammatory Response. Evid. Based Complement. Altern. Med. 2022, 2022, 5752803. [Google Scholar] [CrossRef]
- Chen, J.; Hu, Y.; Chen, L.; Liu, W.; Mu, Y.; Liu, P. The effect and mechanisms of Fuzheng Huayu formula against chronic liver diseases. Biomed. Pharmacother. 2019, 114, 108846. [Google Scholar] [CrossRef]
- Hu, X.Q.; Song, Y.N.; Wu, R.; Cai, F.F.; Zhang, Y.; Peng, J.H.; Hu, Y.Y.; Su, S.B. Metabolic mechanisms of Fuzheng-Huayu formula against liver fibrosis in rats. J. Ethnopharmacol. 2019, 238, 111888. [Google Scholar] [CrossRef]
- Li, X.M.; Peng, J.H.; Sun, Z.L.; Tian, H.J.; Duan, X.H.; Liu, L.; Ma, X.; Feng, Q.; Liu, P.; Hu, Y.Y. Chinese medicine CGA formula ameliorates DMN-induced liver fibrosis in rats via inhibiting MMP2/9, TIMP1/2 and the TGF-beta/Smad signaling pathways. Acta Pharmacol. Sin. 2016, 37, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.D.; Xiao, Z.; Tian, X.T.; Liu, W.; Xu, Z.; Yang, T.; Hu, Y.H.; Zhou, X.X.; Fang, J.; Gao, S.Q.; et al. The Novel Chinese Medicine JY5 Formula Alleviates Hepatic Fibrosis by Inhibiting the Notch Signaling Pathway. Front. Pharmacol. 2021, 12, 671152. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.Y.; Li, W.D.; Liu, C.B.; Jiang, Z.S.; Gu, P.F.; Li, J.Q.; Wang, W.; You, R.L.; Ba, Q.; et al. Rebalancing TGF-beta/Smad7 signaling via Compound kushen injection in hepatic stellate cells protects against liver fibrosis and hepatocarcinogenesis. Clin. Transl. Med. 2021, 11, e410. [Google Scholar] [CrossRef]
- Gong, Z.; Lin, J.; Zheng, J.; Wei, L.; Liu, L.; Peng, Y.; Liang, W.; Hu, G. Dahuang Zhechong pill attenuates CCl4-induced rat liver fibrosis via the PI3K-Akt signaling pathway. J. Cell. Biochem. 2020, 121, 1431–1440. [Google Scholar] [CrossRef]
- Ke, C.; Gao, J.L.; Tu, J.Y.; Wang, Y.; Xiao, Y.X.; Wu, Y.; Liu, Y.J.; Zhou, Z.S. Ganfule capsule alleviates bile duct ligation-induced liver fibrosis in mice by inhibiting glutamine metabolism. Front. Pharmacol. 2022, 13, 930785. [Google Scholar] [CrossRef]
- Bai, T.; Lian, L.H.; Wu, Y.L.; Wan, Y.; Nan, J.X. Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling pathways in activated hepatic stellate cells. Int. Immunopharmacol. 2013, 15, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.J.; Hsu, S.M.; Chien, C.Y.; Wang, Y.H.; Hsu, M.H.; Suk, F.M. Treatment with a New Barbituric Acid Derivative Exerts Antiproliferative and Antimigratory Effects against Sorafenib Resistance in Hepatocellular Carcinoma. Molecules 2020, 25, 2856. [Google Scholar] [CrossRef] [PubMed]
- Sokmen, B.B.; Ugras, S.; Sarikaya, H.Y.; Ugras, H.I.; Yanardag, R. Antibacterial, antiurease, and antioxidant activities of some arylidene barbiturates. Appl. Biochem. Biotechnol. 2013, 171, 2030–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Li, S.; Ma, L.; Cheng, L.; Deng, C.; Chen, Z.; Xie, C.; Xiang, M.; Jiang, W.; Chen, L. A novel agonist of PPAR-gamma based on barbituric acid alleviates the development of non-alcoholic fatty liver disease by regulating adipocytokine expression and preventing insulin resistance. Eur. J. Pharmacol. 2011, 659, 244–251. [Google Scholar] [CrossRef]
- Shen, D.F.; Cheng, H.; Cai, B.Z.; Cai, W.F.; Wang, B.; Zhu, Q.; Wu, Y.B.; Liu, M.; Chen, R.J.; Gao, F.F.; et al. N-n-Butyl haloperidol iodide ameliorates liver fibrosis and hepatic stellate cell activation in mice. Acta Pharmacol. Sin. 2022, 43, 133–145. [Google Scholar] [CrossRef]
- Jiao, W.X.; Bai, M.; Yin, H.W.; Liu, J.Y.; Sun, J.; Su, X.X.; Zeng, H.H.; Wen, J.H. Therapeutic Effects of an Inhibitor of Thioredoxin Reductase on Liver Fibrosis by Inhibiting the Transforming Growth Factor-beta 1/Smads Pathway. Front. Mol. Biosci. 2021, 8, 690170. [Google Scholar] [CrossRef]
- Kong, L.J.; Li, H.; Du, Y.J.; Pei, F.H.; Hu, Y.; Zhao, L.L.; Chen, J. Vatalanib, a tyrosine kinase inhibitor, decreases hepatic fibrosis and sinusoidal capillarization in CCl4-induced fibrotic mice. Mol. Med. Rep. 2017, 15, 2604–2610. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Liu, D.G.; Yang, G.; Kong, L.J.; Du, Y.J.; Wang, H.Y.; Li, F.D.; Pei, F.H.; Song, J.T.; Fan, Y.J.; et al. Endostar, a novel human recombinant endostatin, attenuates liver fibrosis in CCl4-induced mice. Exp. Biol. Med. (Maywood) 2014, 239, 998–1006. [Google Scholar] [CrossRef]
- Song, Z.H.; Liu, X.H.; Zhang, W.; Luo, Y.; Xiao, H.; Liu, Y.; Dai, G.Q.; Hong, J.; Li, A.M. Ruxolitinib suppresses liver fibrosis progression and accelerates fibrosis reversal via selectively targeting Janus kinase 1/2. J. Transl. Med. 2022, 20, 157. [Google Scholar] [CrossRef]
- Lu, Z.N.; Niu, W.X.; Zhang, N.; Ge, M.X.; Bao, Y.Y.; Ren, Y.; Guo, X.L.; He, H.W. Pantoprazole ameliorates liver fibrosis and suppresses hepatic stellate cell activation in bile duct ligation rats by promoting YAP degradation. Acta Pharmacol. Sin. 2021, 42, 1808–1820. [Google Scholar] [CrossRef]
- Yoon, Y.C.; Fang, Z.H.; Lee, J.E.; Park, J.H.; Ryu, J.K.; Jung, K.H.; Hong, S.S. Selonsertib Inhibits Liver Fibrosis via Downregulation of ASK1/MAPK Pathway of Hepatic Stellate Cells. Biomol. Ther. 2020, 28, 527–536. [Google Scholar] [CrossRef]
- Marti-Rodrigo, A.; Alegre, F.; Moragrega, A.B.; Garcia-Garcia, F.; Marti-Rodrigo, P.; Fernandez-Iglesias, A.; Gracia-Sancho, J.; Apostolova, N.; Esplugues, J.V.; Blas-Garcia, A. Rilpivirine attenuates liver fibrosis through selective STAT1-mediated apoptosis in hepatic stellate cells. Gut 2020, 69, 920–932. [Google Scholar] [CrossRef]
- Bisht, S.; Khan, M.A.; Bekhit, M.; Bai, H.; Cornish, T.; Mizuma, M.; Rudek, M.A.; Zhao, M.; Maitra, A.; Ray, B.; et al. A polymeric nanoparticle formulation of curcumin (NanoCurc) ameliorates CCl4-induced hepatic injury and fibrosis through reduction of pro-inflammatory cytokines and stellate cell activation. Lab. Investig. 2011, 91, 1383–1395. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Cao, J.; Chen, M.; Zhang, Y. Recent advances in the role of exosomes in liver fibrosis. J. Gastroenterol. Hepatol. 2023. [Google Scholar] [CrossRef]
- Wang, N.; Li, X.; Zhong, Z.; Qiu, Y.; Liu, S.; Wu, H.; Tang, X.; Chen, C.; Fu, Y.; Chen, Q.; et al. 3D hESC exosomes enriched with miR-6766-3p ameliorates liver fibrosis by attenuating activated stellate cells through targeting the TGFbetaRII-SMADS pathway. J. Nanobiotechnol. 2021, 19, 437. [Google Scholar] [CrossRef]
- Tang, M.; Guo, C.; Sun, M.; Zhou, H.; Peng, X.; Dai, J.; Ding, Q.; Wang, Y.; Yang, C. Effective delivery of osteopontin small interference RNA using exosomes suppresses liver fibrosis via TGF-beta1 signaling. Front. Pharmacol. 2022, 13, 882243. [Google Scholar] [CrossRef]
- Song, Z.; Chen, W.; Athavale, D.; Ge, X.; Desert, R.; Das, S.; Han, H.; Nieto, N. Osteopontin Takes Center Stage in Chronic Liver Disease. Hepatology 2021, 73, 1594–1608. [Google Scholar] [CrossRef]
- Poilil Surendran, S.; George Thomas, R.; Moon, M.J.; Jeong, Y.Y. Nanoparticles for the treatment of liver fibrosis. Int. J. Nanomed. 2017, 12, 6997–7006. [Google Scholar] [CrossRef] [Green Version]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug. Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, M.; Fu, L.H.; Lin, J.; Zhou, X.M.; Zhou, P.H.; Huang, P.; Hu, H.; Han, Y. Liver-targeted delivery of TSG-6 by calcium phosphate nanoparticles for the management of liver fibrosis. Theranostics 2020, 10, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, S.A.; Chitkara, D.; Mahato, R.I.; Mittal, A. Lipid based nanocarriers for effective drug delivery and treatment of diabetes associated liver fibrosis. Adv. Drug Deliv. Rev. 2021, 173, 394–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Byun, J.; Shim, G.; Oh, Y.K. Fibroblast activation protein activated antifibrotic peptide delivery attenuates fibrosis in mouse models of liver fibrosis. Nat. Commun. 2022, 13, 1516. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Okimoto, S.; Kuroda, S.; Tashiro, H.; Kobayashi, T.; Taogoshi, T.; Matsuo, H.; Ohdan, H. Vitamin A-coupled liposomal Rho-kinase inhibitor ameliorates liver fibrosis without systemic adverse effects. Hepatol. Res. 2019, 49, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, R.; Liu, Q.; Zhou, J.; Huang, H.; Huang, Y.; Zhang, Z.; Wu, T.; Tang, Q.; Huang, C.; et al. SB431542-Loaded Liposomes Alleviate Liver Fibrosis by Suppressing TGF-beta Signaling. Mol. Pharm. 2020, 17, 4152–4162. [Google Scholar] [CrossRef]


| Compounds | Animal Model | Targets/Pathways/Mechanisms | Reference |
|---|---|---|---|
| Pien-Tze-Huang (PZH) | CCl4-induced liver fibrosis in male SD rats | Autophagy and TGF-β1/Smad2 signaling pathways | [80] |
| Inhibiting HSC activation and diminishing fibrogenesis | |||
| Compound Kushen injection (CKI) | CCl4-induced chronic liver fibrosis in C57BL/6 mice | Inhibiting HSC activation | [138] |
| MCD diet-induced NASH model in C57BL/6 mice | Rebalancing TGF-β/Smad7 signaling | ||
| CCl4-induced HCC models in C57BL/6 mice | |||
| JY5 | CCl4-induced liver fibrosis in male Wistar or SD rats | Inhibition of the Notch signaling pathway | [137] |
| BDL-induced liver fibrosis in male Wistar or SD rats | Inhibiting the activation of HSCs | ||
| CCl4-induced liver fibrosis in male C57/BL6 mice | |||
| Pokeweed antiviral protein (PAP) | CCl4-induced liver fibrosis in male C57/BL6 mice | Inhibition of glycolysis through regulation of the Wnt/JNK pathway | [24] |
| Suppressing HSC activation | |||
| San-Cao Granule (SCG) | TAA-induced liver fibrosis in SD rats | Inhibiting the TGF-β1/Smad pathway | [132] |
| CGA | DMN-induced liver fibrosis in male Wistar rats | Downregulation of MMP2/9 activities and TIMP1/2 protein expression | [136] |
| Inhibition of the TGF-β1/Smad signaling pathway | |||
| Inhibiting the activation of HSCs | |||
| Dahuang Zhechong pill (DHZCP) | CCl4-induced liver fibrosis in male SD rats | Inactivating the PI3K/Akt pathway | [139] |
| Suppressing HSC proliferation | |||
| Jageum–Jung (JGJ) | CCl4-induced liver fibrosis in male C57/BL6 mice | Inhibiting the activation and translocation of STAT-1 and NF-κB | [109] |
| Regulating TGF-β1/Smad signaling. | |||
| Inhibiting HSC activation | |||
| Ganfule capsule (GFL) | BDL-induced liver fibrosis in male C57/BL6 mice | Inhibiting glutamine metabolism | [140] |
| Restricting the activation of the NF-κB pathway |
| Compounds | Animal Model | Targets/Pathways/Mechanisms | Reference |
|---|---|---|---|
| N-n-Butyl haloperidol iodide (F2) | TAA-induced liver fibrosis in C57BL/6J mice | Reducing responsiveness of HSCs to TGF-β1 | [146] |
| CCl4-induced liver fibrosis in C57BL/6J mice | Inhibiting activated HSCs | ||
| Butaselen (BS) | CCl4-induced liver fibrosis in male BALB/c mice | Inducing the apoptosis of activated HSCs | [147] |
| Inhibiting the production of α-SMA and collagens by HSCs | |||
| Downregulating TGF-β1 expression and blocking the TGF-β1/Smads pathway | |||
| barbituric acid (BA)-5 | CCl4-induced liver fibrosis in male C57BL/6 mice | Blocking both the TGF-β1 and LPS-induced NF-κB signaling pathways | [102] |
| Inhibiting HSC activation and liver fibrosis | |||
| Inhibiting macrophages recruitment and activation | |||
| JD5037 | CCl4-induced liver fibrosis in male mice | Blocking the CB1 receptor/β-arrestin1/Akt signaling pathway | [79] |
| BDL-induced liver fibrosis in male mice | Blocking the activation of HSCs | ||
| GNS561 | DEN-induced liver fibrosis in male rats | Disrupting TGF-β1 maturation and TGF-β1/Smad and MAPK signaling | [16] |
| Inducing the apoptosis of HSCs | |||
| Preventing HSC activation and decreasing ECM deposition | |||
| Cultured bear bile powder (CBBP) | DMN-induced liver fibrosis in wild-type Wistar rats | Elevating the expression of PPAR-α and PPAR-γ | [69] |
| Improving β-FAO and inhibiting inflammation | |||
| Vatalanib | CCl4-induced liver fibrosis in male BALB/c mice | Reducing liver inflammation | [148] |
| Endostar | CCl4-induced liver fibrosis in male BALB/c mice | Improving liver function and reducing liver inflammation | [149] |
| Inhibiting a-SMA protein expression | |||
| Reducing collagen accumulation | |||
| HS-173 | CCl4-induced liver fibrosis in male BALB/c mice | Blocking the PI3K/Akt pathway | [142] |
| Promoting HSC apoptosis | |||
| Inhibiting the expression of fibrotic mediators | |||
| Ruxolitinib | CCl4-induced liver fibrosis in male C57BL/6 mice | Blocking JAK1/2 pathway | [150] |
| TAA-induced liver fibrosis in male C57BL/6 mice | Inhibiting the activation of HSCs | ||
| Pantoprazole (PPZ) | BDL-induced liver fibrosis in male SD rats | Promoting the proteasome-dependent degradation and ubiquitination of YAP | [151] |
| Inhibiting HSC activation | |||
| Disrupting the interaction between OTUB2 and YAP | |||
| Tenofovir disoproxil fumarate (TDF) | TAA-induced liver fibrosis in male C57BL/6 mice | Downregulating the PI3K/Akt/mTOR signaling pathway | [96] |
| The apoptosis of activated HSCs | |||
| Selonsertib | DMN-induced liver fibrosis in male SD rats | Blocking ASK1/MAPK signaling | [152] |
| Inducing apoptosis of HSCs | |||
| Rilpivirine (RPV) | CCl4-induced fibrosis in female C57BL/6J mice | Selective STAT1-dependent induction of apoptosis in HSCs | [153] |
| BDL-induced fibrosis in female C57BL/6J mice | Promoting liver regeneration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Q.; Yi, Q.; Tang, L. Liver Fibrosis Resolution: From Molecular Mechanisms to Therapeutic Opportunities. Int. J. Mol. Sci. 2023, 24, 9671. https://doi.org/10.3390/ijms24119671
Pei Q, Yi Q, Tang L. Liver Fibrosis Resolution: From Molecular Mechanisms to Therapeutic Opportunities. International Journal of Molecular Sciences. 2023; 24(11):9671. https://doi.org/10.3390/ijms24119671
Chicago/Turabian StylePei, Qiying, Qian Yi, and Liling Tang. 2023. "Liver Fibrosis Resolution: From Molecular Mechanisms to Therapeutic Opportunities" International Journal of Molecular Sciences 24, no. 11: 9671. https://doi.org/10.3390/ijms24119671