1. Introduction
Cervical Spondylotic Myelopathy (CSM) is the most common cervical spinal cord dysfunction in middle-aged and elderly people. Vascular dysfunction and neural impairment have been demonstrated to be a crucial pathophysiological process in chronic compressive SCI [
1,
2]. Surrounding degenerative tissue, inducing intermittent or continuous compression to the spinal cord, which cause ischemia, disruption of blood spinal cord barrier (BSCB), neuronal apoptosis and damage of descending or ascending axon, eventually lead to incomplete neurological deficit [
3,
4]. Although it has been widely believed that neural regeneration is extremely limited in complete SCI of adult mammals, spontaneous functional recovery (SFR) commonly occurs in incomplete SCI [
5,
6,
7]. Restoration of BSCB integrity, neuroprotective effects by neurotrophic factor secretion, axonal remyelination by oligodendrocyte precursors and axonal remodeling such as axonal sprout formation may contribute to spontaneous recovery after incomplete SCI [
7,
8].
The neurovascular unit (NVU), integrated by neurons, microvascular endothelial cells and the surrounding neuroglial cells, i.e., oligodendrocytes and astrocytes, constitutes a fundamental functional unit to enable intercellular communication and signaling transduction [
9,
10]. The highly specified monolayer endothelial cells with the perivascular pericytes and the surrounding astrocytes (astrocytic endfeet) constitute the integrated BSCB, which plays an essential role in regulating blood supply and substances exchange, thus maintaining microenvironment homeostasis of CNS [
11,
12]. The osculating connection facilitates cell to cell interaction among NVU compartments. Interactions between endothelial cells and astrocytes maintain the functional integrity of the BSCB [
11]. Therefore, it is necessary to reveal the association change among components of NVU and fully elucidate the pathophysiological mechanism of this association in the natural process of SFR, which could help to shed light on the promising therapeutic strategy of CSM.
Although numerous studies have demonstrated that dysfunction or disruption of NVU is the main pathophysiological change response for ischemic cerebral insult or degenerative central nervous system (CNS) disorders [
10,
13,
14,
15], the existing evidence regarding spontaneous NVU remodeling in the structural and functional recovery of SCI is extremely limited. A recent study has revealed several degenerative ultrastructural changes of NVU including degeneration of neurons and axons, vacuolation of endothelium, breakdown of the tight junction and swelling of astrocytic endfeet and mitochondria in chronic compressive cervical SCI [
16]. Compensatory changes of vascular components including proliferation of endothelium, thickening of basement membrane and increment of pericyte vessel coverage were observed in the compressive epicenter [
16]. However, the pathomechanism of NVU compensation contributing to SFR has not been elaborated.
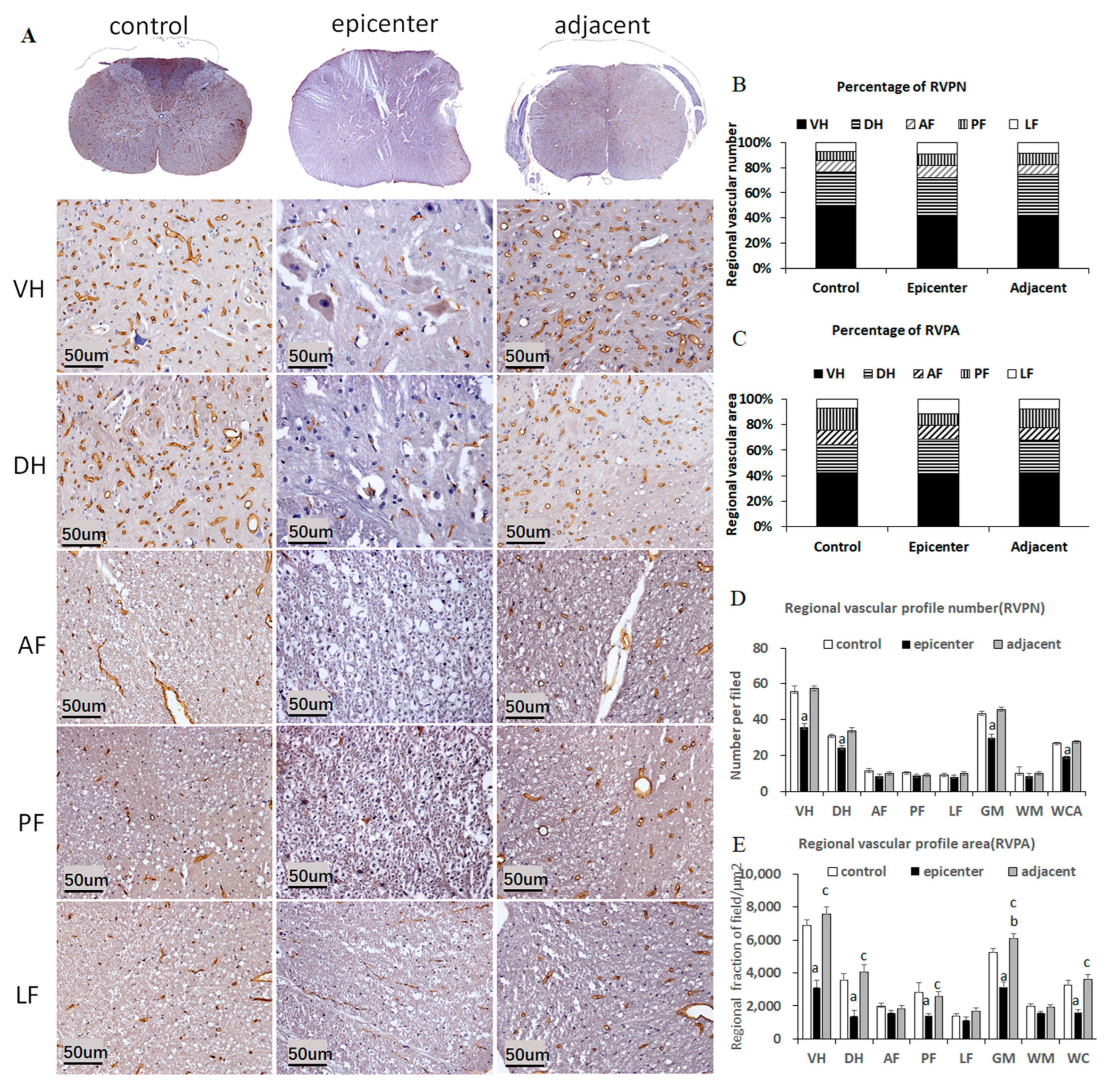
As a specific marker for CNS blood vessels, endothelial barrier antigen (EBA) was used to detect the integrity of the BSCB [
17,
18]. The present study was an elementary attempt to disclose pathophysiological changes in each NVU component, in particular in the adjacent level, in an experimental CSM model. Ultrastructural features of NVU components were revealed by Transmission Electron Microscopy (TEM). Evan blue (EB) extravasation testing was used to detect the permeability of the BSCB. EBA-immunoreactivity was used for blood vessel quantitative analysis. Neuroglial cells including astrocytes, oligodendrocytes and microglial cells were also specifically visualized by immunohistochemical staining. The interrelation among quantity of microvessel and neuroglial cells were analyzed.
3. Discussion
The clinical presentation of CSM varies with a big spectrum of spontaneous progression. Some patients with CSM showed linear deterioration in neurological function. Some patients present stable disability followed by episodes of deterioration, while others showed a steady disability to a sudden decline. Currently, surgical decompression is the mainstay treatment strategy of CSM. However, the efficacy of neurological recovery following surgical decompression appears to be a large uncertainty, i.e., some patients achieve satisfactory recovery, while others remain residually disabled, even showing unpredictable deterioration. The pathomechanism underlying such existing clinical contradictions remains poorly understood. Limited postmortem evidence has proved neural (neuron and oligodendrocyte) apoptosis, inflammatory responses, glial scar formation, neuronal degeneration/loss and axonal demyelination as the most prominent pathophysiological features for CSM [
19]. In the present study, we have undertaken an investigation of the NVU changes along the natural course of SFR. We found that the compensatory change of NVU, in particular in the adjacent level of the compressive epicenter, was involved in functional recovery in experimental CSM model. This implies the effect of NVU compensation from the adjacent level of the myelopathic cervical cord on functional recovery.
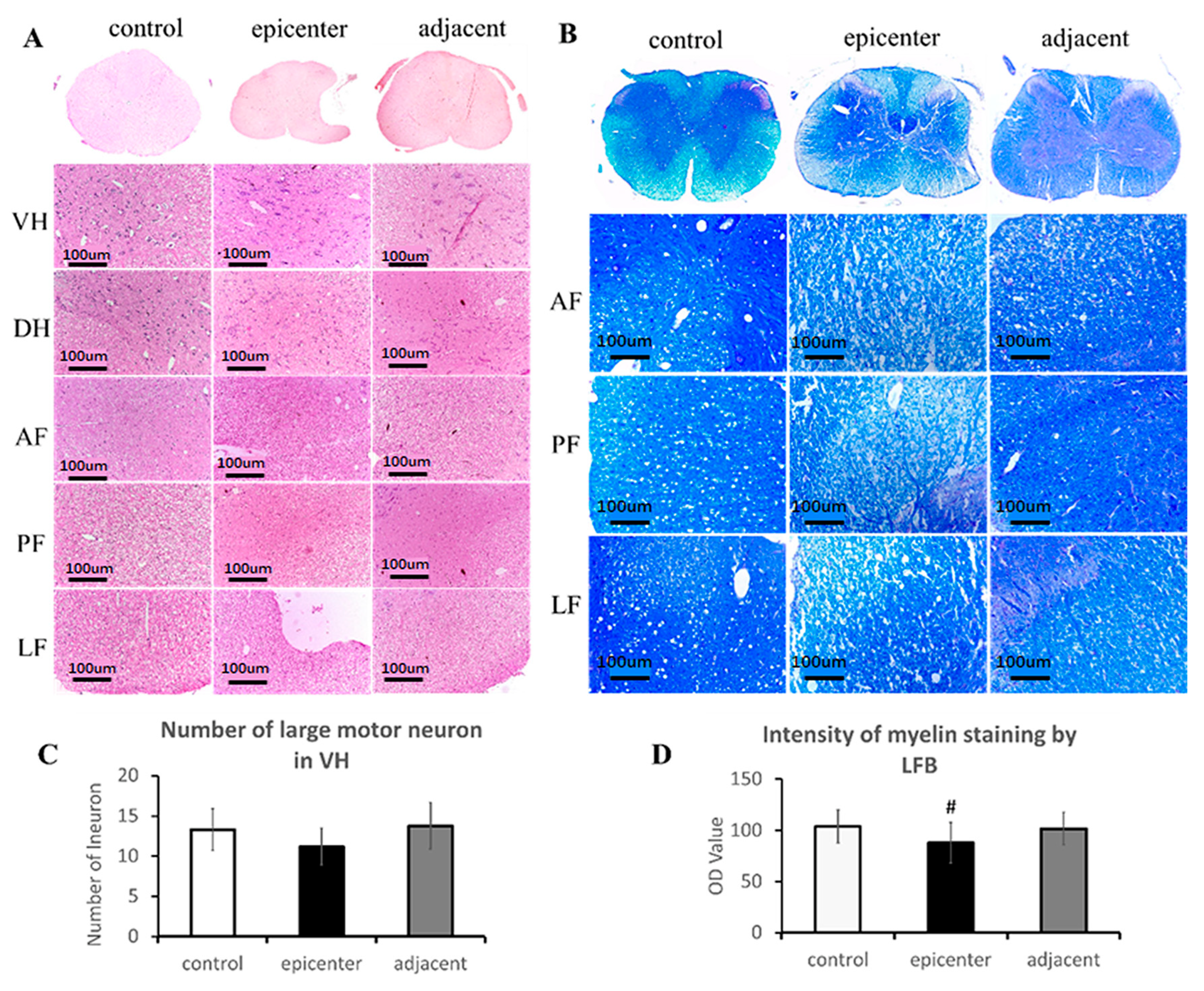
The primary neuropathological features of CSM, which were well documented in postmortem CSM patient and clinically relevant models of CSM, include degeneration and loss of neuron in the ventral horn and loss of axon and demyelination in the posterior and lateral column of WM [
4,
19,
20,
21]. Agreeing with previous studies, the present study found significant loss of neurons and axons in the compressive epicenter but not in the adjacent level. Neuronal and axonal degeneration were verified by pathological and ultrastructural examination in the compressive epicenter, as well as with mild degenerative change in the adjacent level. Although neuronal and axonal degenerative change existed throughout the long-term compressive condition, it has to be noted that synaptic compensatory changes, in particular, such as increased synaptic vesicle and reactive synaptic mitochondria, were supposed to play an important role in compensatory pathomechanisms, which are responsible for spontaneous functional recovery. Our ultrastructural findings further strengthen the underlying pathomechanism that surgical decompression induced restoration of serotonergic fibers as one of endogenous mechanisms for promoting neurological recovery [
22].
The NVU, mainly composed of the BSCB and neurons, is the essential functional unit of the CNS, which plays an important part in maintaining the integrity of the BSCB and regulating the permeability of the BSCB and microcirculation [
9,
23]. Structural disruptions of the BSCB commonly involve endothelial dysfunction/injury, tight junction breakdown, basement membrane corrosion and astrocytic endfeet detachment from the abluminal wall of capillaries [
16]. The disappearance of EBA-immunoreactivity was reported to be correlated with tissues edema, inflammatory condition and astrocytic swelling [
24,
25]. On the contrary, the reappearance may be associated with restoration of the BSCB property benefitting from the surrounding astrocytic endfeet and compensatory proliferation of endothelial cells [
26]. Loss of EBA immunoreactivity was documented in a period of approximately 2 weeks in the injury focal point, as well as in the adjacent microvessels in acute brain injury [
24], while gradual restoration of EBA immunoreactivity was also observed within 3–4 weeks [
18], which confirms that the endothelium, as one of the essential components of the NVU, plays a vital role in the pathophysiological process of CNS injury and recovery [
12]. Further studies described significant decrement of immunostaining intensity, vascular profile number and area in the contusion and pericontusional site of the traumatic brain cortical, providing a quantitative method for exploring the microvascular alterations [
25,
27]. However, the correlation between morphological and quantitative change of EBA-positive microvessels and neurological functions was not carried out in these studies. In a traumatic SCI model, an earlier reappearance of EBA-immunoreactivity was found at the compressive site after 9 days thoracic cord compression [
26]. However, the pathophysiological mechanism underlying reappearance of EBA-positive
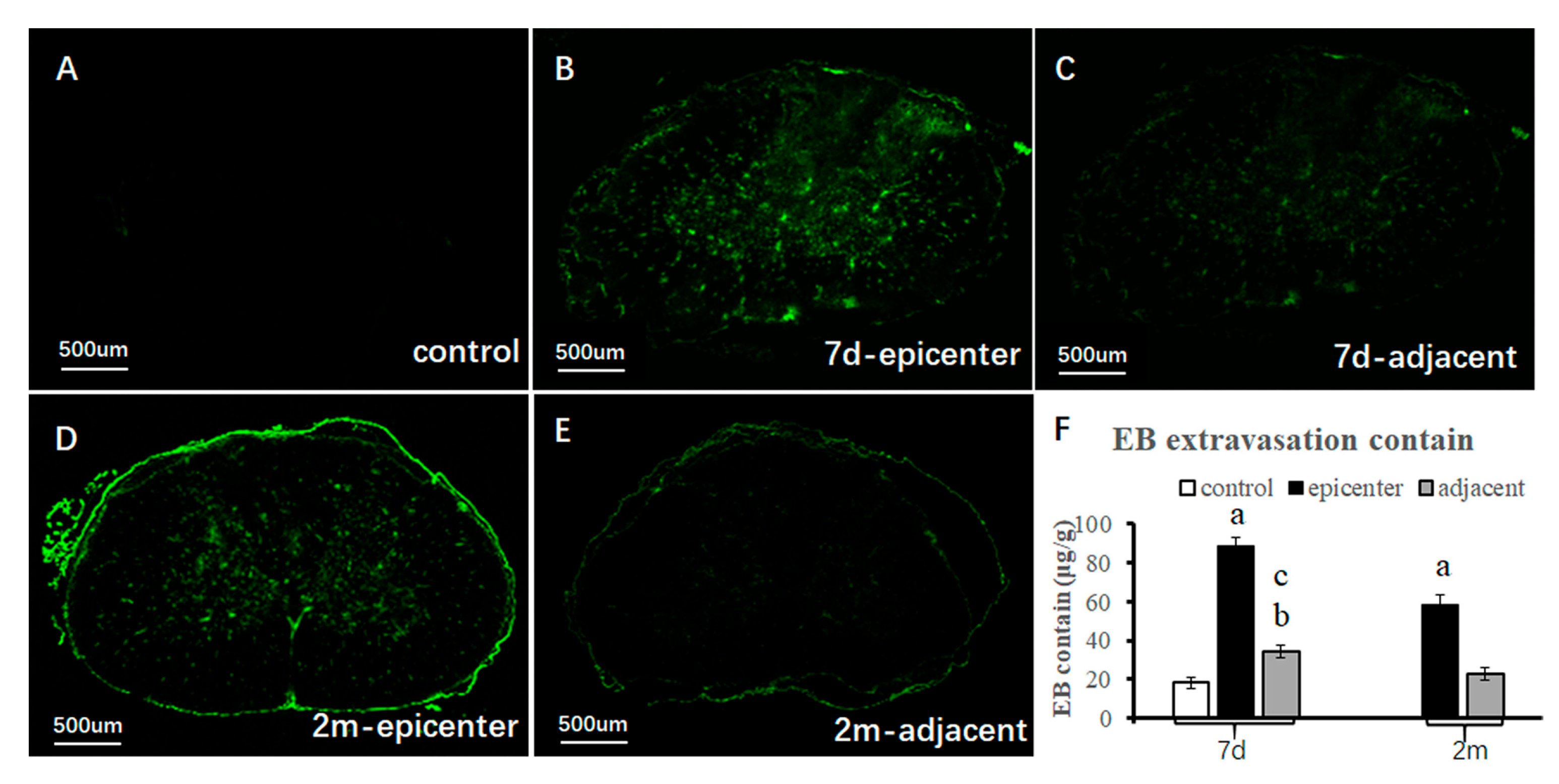
microvessels and the corresponding functional recovery remains unclear. In contrast with traumatic CSN injury, our aforementioned findings exhibited long-term loss of EBA immunoreactivity and reduction of RVPA in the compressive epicenter. The morphological characteristics of the EBA immunolabeling microvessel were quite consistent with the ultrastructural features by TEM examination. Furthermore, outstanding restoration of such changes was found in the adjacent level. The quantitative findings, to some extent, indicated dysfunction of microcirculation in the compressive site, while the compensatory change of the endothelial barrier in the adjacent level was an important compensatory change for improving microcirculation and thus inducing functional recovery. The pathophysiological change different from traumatic or acute CNS injury should be responsible for the unique pathogenic mechanism that persistent cord compression poses as long-term oppression to the microvessel [
3,
4]. In consequence, these findings indicated that compensatory restoration of endothelial barrier function and microcirculation may be one of the endogenous driving forces for inducing SFR in a CSM model.
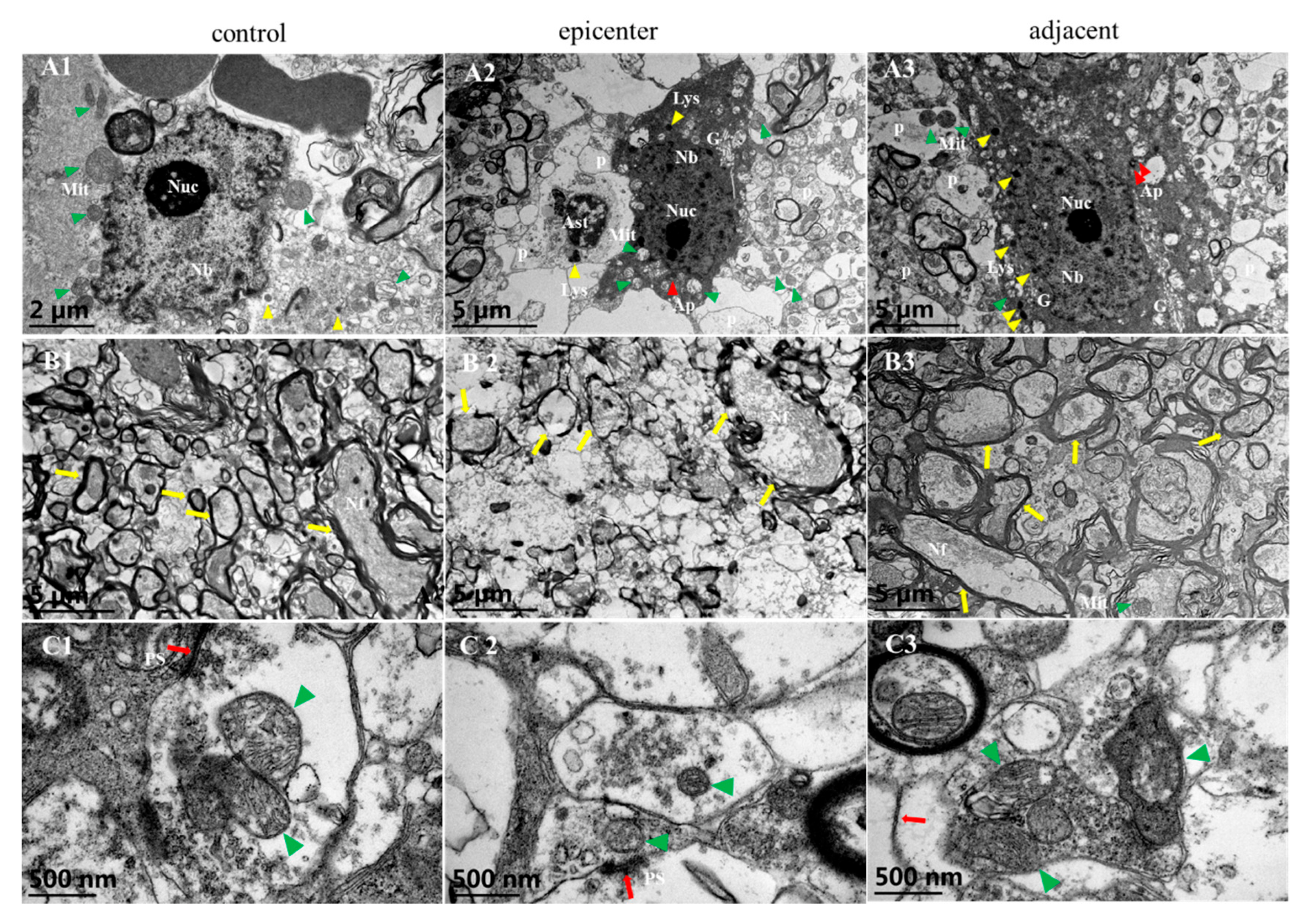
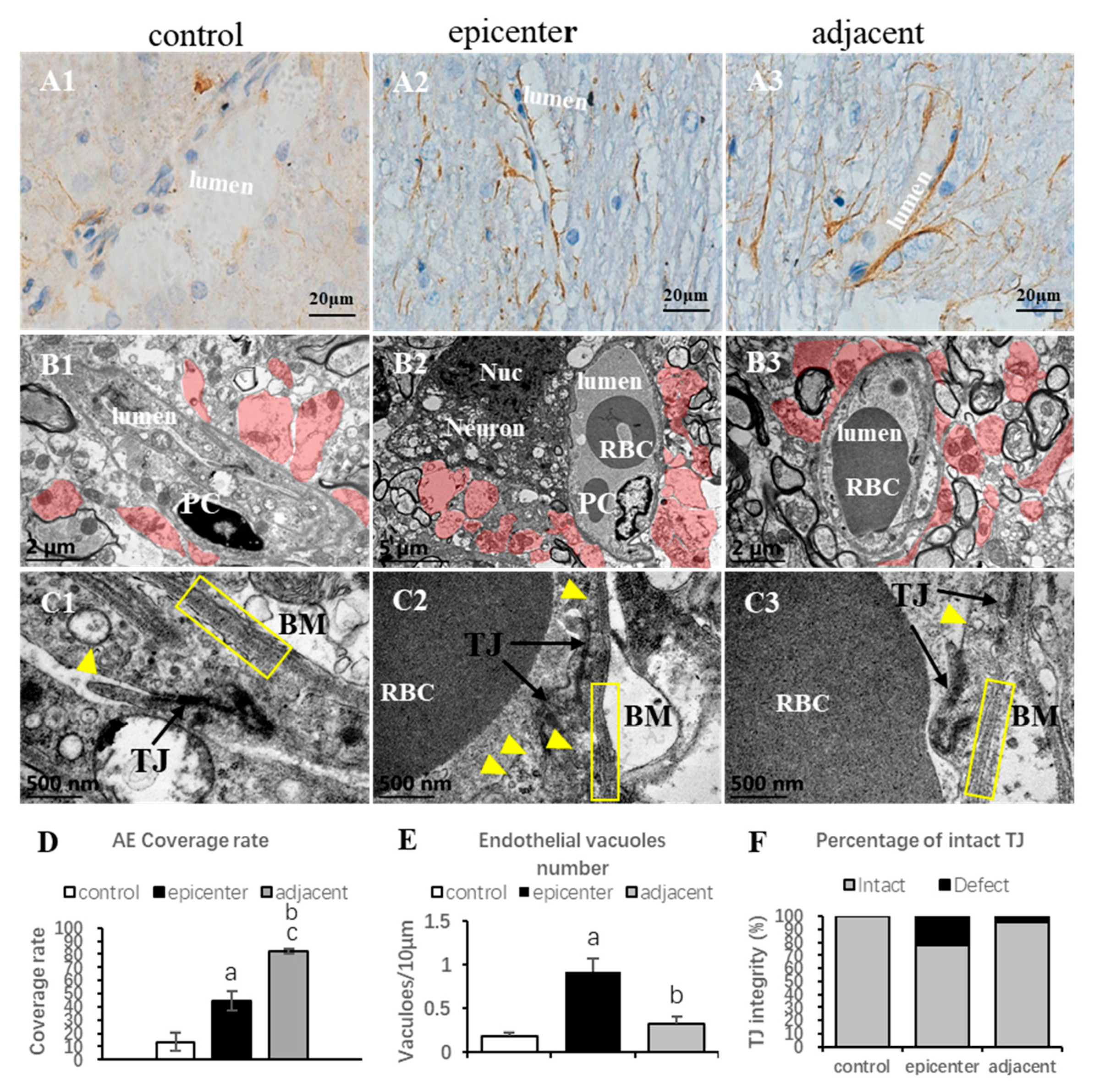
Our TEM examination further proved that remarked vacuolation of endothelial cells which increased the defect of integrity and fuzzy structure of basement membrane were the ultrastructural characteristics of microvascular destruction in the compressive epicenter. Meanwhile, proliferation of astrocytic endfeet was more apparent in the adjacent level than that in the compressive epicenter. Vascular events, in particular, disruption of the BSCB, are usually the early pathophysiological change in SCI [
28]. In the natural course of CSM, progressive chronic compression, inducing ischemia and subsequent disruption of BSCB, are considered to trigger a cascade of pathophysiological processes, including oxidative stress damage, neuroinflammation and neurotoxicity impairment, which finally lead to neuronal degeneration/apoptosis and axonal demyelination [
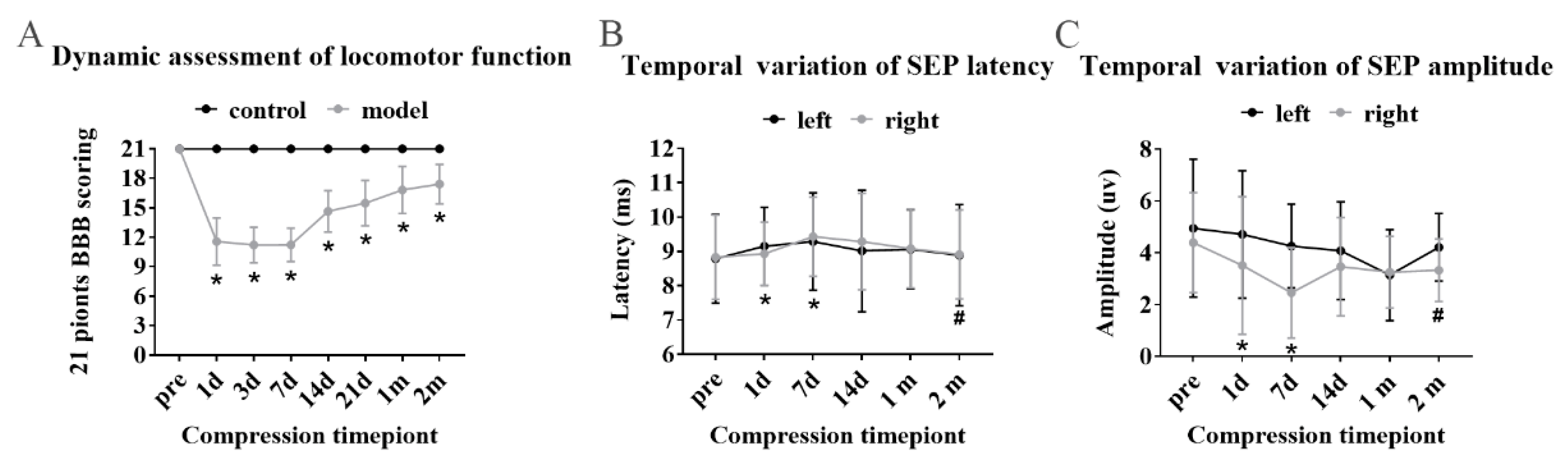
29]. The latest research has proved that increased permeability and disruption of the BSCB are presented in patients with degenerative cervical myelopathy, of which damage magnitude was correlated with the severity of neurological symptoms [
2]. In this study, we further validated that functional disruption of BSCB integrity was synchronized with neurological deficit in the early stage from the 1st day to 7th day of cord compression. From the 14th day to the observation termination period at the 2nd month, gradual SFR was documented with functional restoration of BSCB integrity. However, at the observation termination, EBA-immunoreactive microvessels showed a significant loss of immunoreactivity and decrement of RVPA in the compressive epicenter, which suggested persistent endothelial barrier dysfunction due to long-term compression in the compressive focal point. Meanwhile, in the adjacent level, an obvious restoration of immunoreactivity and increment of RVPA was seen, especially in the VH of GM. A similar chronic vascular change documented in a rare postmortem CSM patient study, which was characterized by abnormal thickening of the vasculature, might suggest that compensatory vasculature responds in long-term compressive conditions [
19].
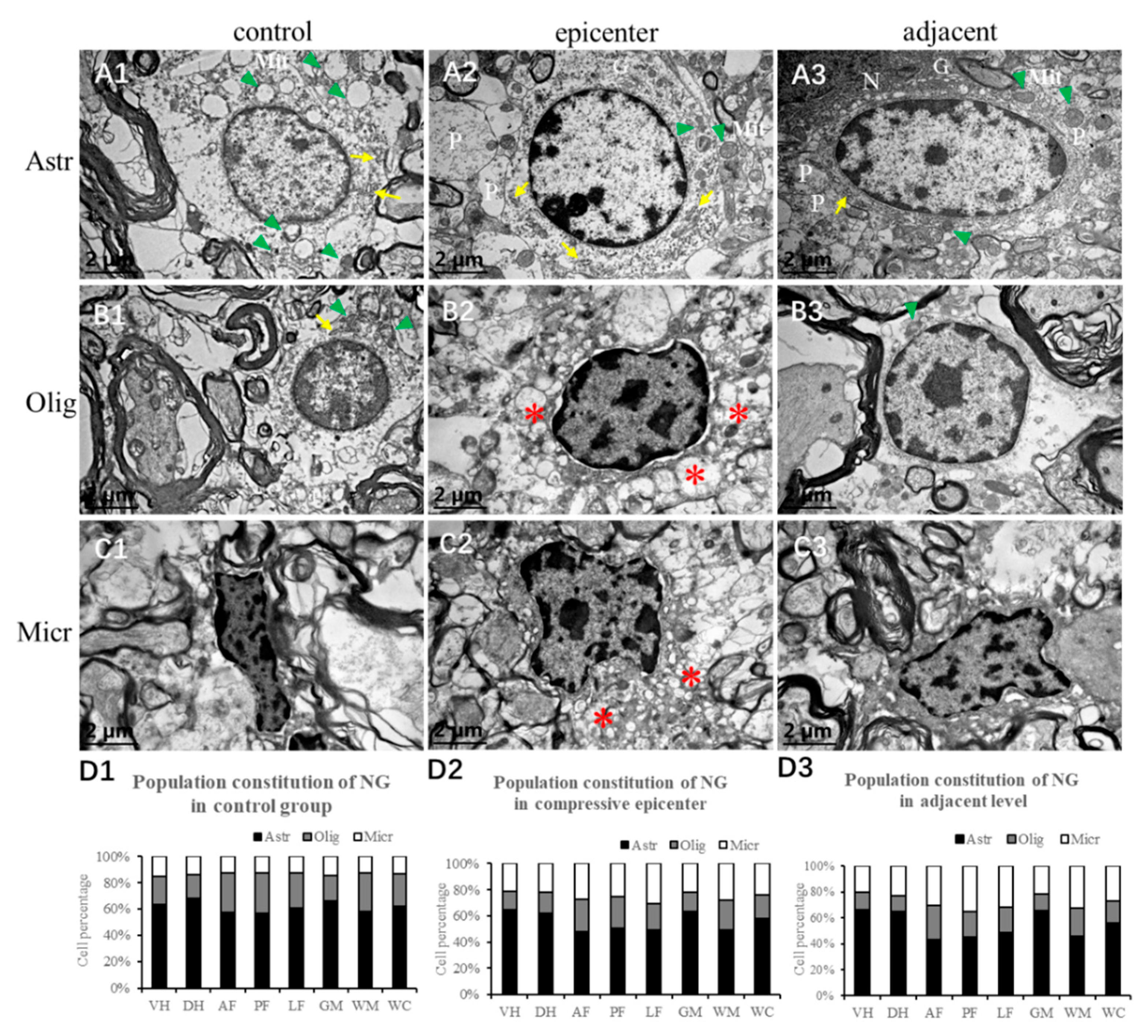
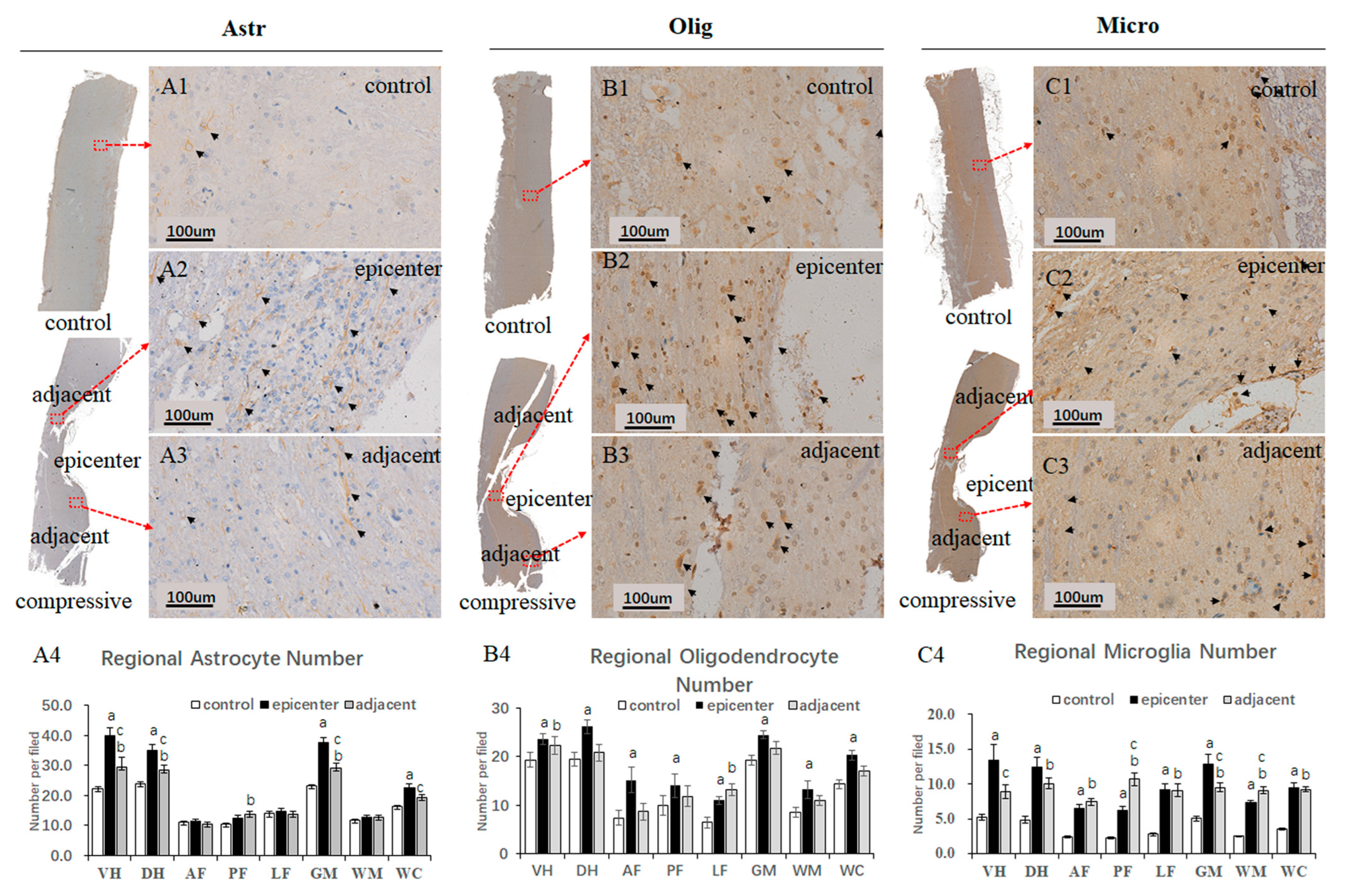
In the present study, conspicuous accumulation of gemistocytic astrocytes with proliferated elongated processes was identified in the compressive epicenter, which was different from the glial scar formation in acute SCI [
30]. The number of astrocytes, oligodendrocytes and microglial cells increased significantly in the compressive epicenter compared to that in the control group. Reactive proliferation and aggregation change of neuroglial cells were related more mildly in the adjacent level, while drastic increment of microglial cells can be seen both in the compressive epicenter and the adjacent level. A growing number of studies have focused on the pathophysiological responses of neuroglial cells, which are supposed to be involved in natural process of CSM and the pathophysiological process of neurorestoration. Decreased myelin content with dysfunction of spinal cord conduction has been verified in a CSM patient [
31]. Since oligodendrocytes are the key neuroglial cells responsible to the production of myelin sheaths and the self-repairing capability of myelin, apoptosis of oligodendrocytes has been implicated as one of essential pathomechanisms in CSM [
19,
32,
33,
34,
35]. However, on the contrary, one of most recent studies has found that spontaneous locomotor recovery was not affected when the axons failed to remyelinate, which suggests that oligodendrocyte remyelination is non-essential for spontaneous recovery [
36]. This conclusion may partially explain why spontaneous recovery occurred regardless of the existence of long-term axonal demyelination in the compressive epicenter in the present study. Therefore, whether axonal remyelination and subsequent functional recovery are ascribed to conspicuous increment of oligodendrocytes in the epicenter and adjacent level needs further validation.
Ultrastructural pathological lesions of astrocytes were documented in autopsied CSM patients [
37]. It has been widely believed that overreactive astrocytes and astrocytic scars were considered as detrimental factors that prevent axonal regrowth [
38,
39]. Nevertheless, a recent study broke the prevailing view and drew the conclusion that astrocytic scar formation is beneficial for axonal regeneration [
40]. Similarly, during the spontaneous recovery process, large number of activated astrocytes characterized by long thorn-shape processes presented in the epicenter as well as the adjacent level, which would prompt us to revisit the role of astrocytes in chronic compressive SCI.
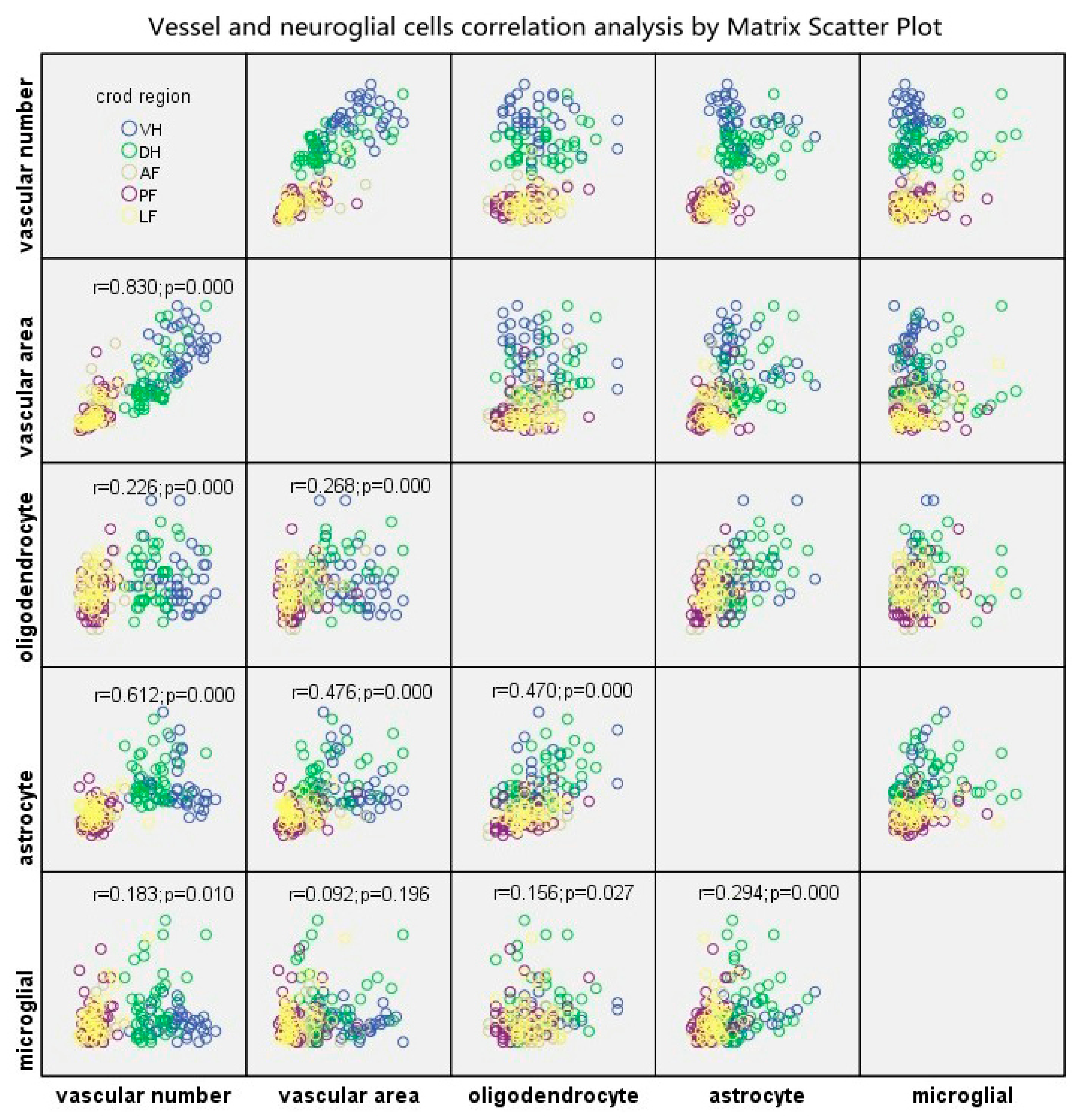
The BSCB, neurons and neuroglial cells are the indispensable components of NVU, and are closely interrelated with each other structurally and functionally, thus, maintaining homeostasis of the CNS. In this present study, The Matrix Scatter Plot disclosed a moderate correlation between astrocyte number and vascular number/vascular area, as well as number of astrocytes and oligodendrocytes. Proliferated astrocytic endfeet also appeared in the epicenter and the adjacent level. Moreover, the disruption of the endothelial barrier was characterized by increased perivascular space and detached astrocyte endfeet in the compressive epicenter. In contrast, the perivascular astrocytic endfeet process was tightly wrapped with the abluminal vascular wall. Such a pathomorphological change of astrocyte–endothelial construction reacting to persistent compression, to some extent, indicated that the astrocytes struggle to maintain the integrity of the BSCB and prevent BSCB disruption spreading to the adjacent level [
11,
14]. The latest study further validates that astrocytes play an important part in regulating microcirculation in the brain and thus modulating the activity of neurons [
41]. Homeostasis interaction among neuroglia, including oligodendrocytes, astrocytes and microglia enable the process of remyelination [
42]. However, abnormal aggregation of perivascular oligodendrocyte precursor cells might disturb the astrocytic endfeet and endothelial tight junction integrity, which may disrupt the BBB, increase vascular permeability and induce inflammatory impairment [
43].
In the adjacent level, we observed evident increased EBA-immunoreactive microvasculature number and area, with closely wrapping proliferated astrocytic endfeet and restoration of BSCB permeability, which may suggest that reactive astrocytes play an important role in maintaining the structural and functional integrity of the BSCB. The compensatory change of microvessels would contribute to rebalancing the microcirculation and promoting neuron survival and synaptic plasticity. An up-to-date study indicated that astrocytes play an important role in regulating cerebral perfusion and circulation condition to maintain sufficient bloody supply [
41]. In addition, the number of astrocytes, oligodendrocytes and microglia increased significantly in the adjacent level, which may be associated with clearance of degeneration or damaged axon and axonal plasticity around the lesion site [
42]. Nonetheless, in brief, the neurovascular compensatory responses, instead of consequential neural regeneration, originating from the spared cord adjacent to the mechanical compressive epicenter, play a crucial role in the natural process of spontaneous recovery of the CSM model.
The NVU has become a hotspot concept for investigating pathophysiological mechanisms of cerebrovascular disease (including ischemia cerebral infarction or hemorrhage) and neurodegeneration disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) [
14]. A recent study revealed the ultrastructural damage to the NVU components in a rat model, including evident endothelium vacuoles, defects of tight junctions, an expansive basement membrane, swollen astrocytic endfeet and mitochondria [
16]. This study also specifically pointed out compensatory ultrastructural changes of microvascular components, which are characterized by increased thickening of the endothelium, an expansive basement membrane, increased pericyte processing area and vessel coverage [
16], while correlation analysis between compensatory change (particularly in the adjacent level) and spontaneous functional recovery in neuroglia and microvessels was not performed. To develop a drug therapeutic mechanism and effect in the NVU level, further attempts have established similar in vitro modeling of the NVU by co-culturing of endothelial cells, neural stem cells, astrocytes and pericytes, [
44,
45]. The latest therapeutic experimental study has obtained evidence on NVU component repairing and the consequent neurobehavior recovery in traumatic cervical SCI [
46]. In consequence, the compensatory response to chronic compression in the NVU level may play a crucial role in spontaneous recovery in CSM.
The present study provided a general observation of the effect of NVU compensation changes during SFR of CSM. Findings of this study will be beneficial for new therapeutic strategy development of CSM. It should be noted that further study in cell biology and fundamental neurological studies are necessary to explain the underlying mechanisms of spontaneous recovery. It also helps to consider diagnostic and prognostic value in neurovascular measurement of the cervical spinal cord. A non-invasive way for spinal cord ultrastructure measurement of is one of the key issues in achieving accurate assessment and precise predictability of CSM. Advanced neuroimaging technology, such as diffusion tensor imaging (DTI) [
47,
48], could provide clear visualization and quantitative assessment of neural deficits in the spinal cord. It would be a promising tool for in vivo exploration of spinal cord ultrastructural features.
4. Materials and Methods
4.1. Animal Models
4.1.1. Compression Material
A water-absorbing and progressive expanding synthetic polyurethane polymer sheet was used as the implanting compression material. The polyurethane polymer sheet was made of isocyanates and polyols (Guangzhou Fischer Chemical Co., Ltd., Guangzhou, China). A polymer sheet at size of 3 mm × 1 mm × 1 mm was cut and sterilized for implantation preparation. The compression sheet could absorb liquid in the epidural space to gradually expand its maximum volume and induced progressive compression to the spinal cord [
49].
4.1.2. Surgical Procedure
A total of 24 female adult Sprague–Dawley (SD) rats (250–300 g) were divided into compressive group for 2 months (n = 12) and control group (n = 12). All animal protocols were approved by the animal ethics committee of the affiliated hospital of Guangdong Medical University, Zhanjiang, Guangdong, China.
The animals were operated on under microscopy by a trained spine surgeon according to an established surgical protocol for implantation of water-absorbing materials [
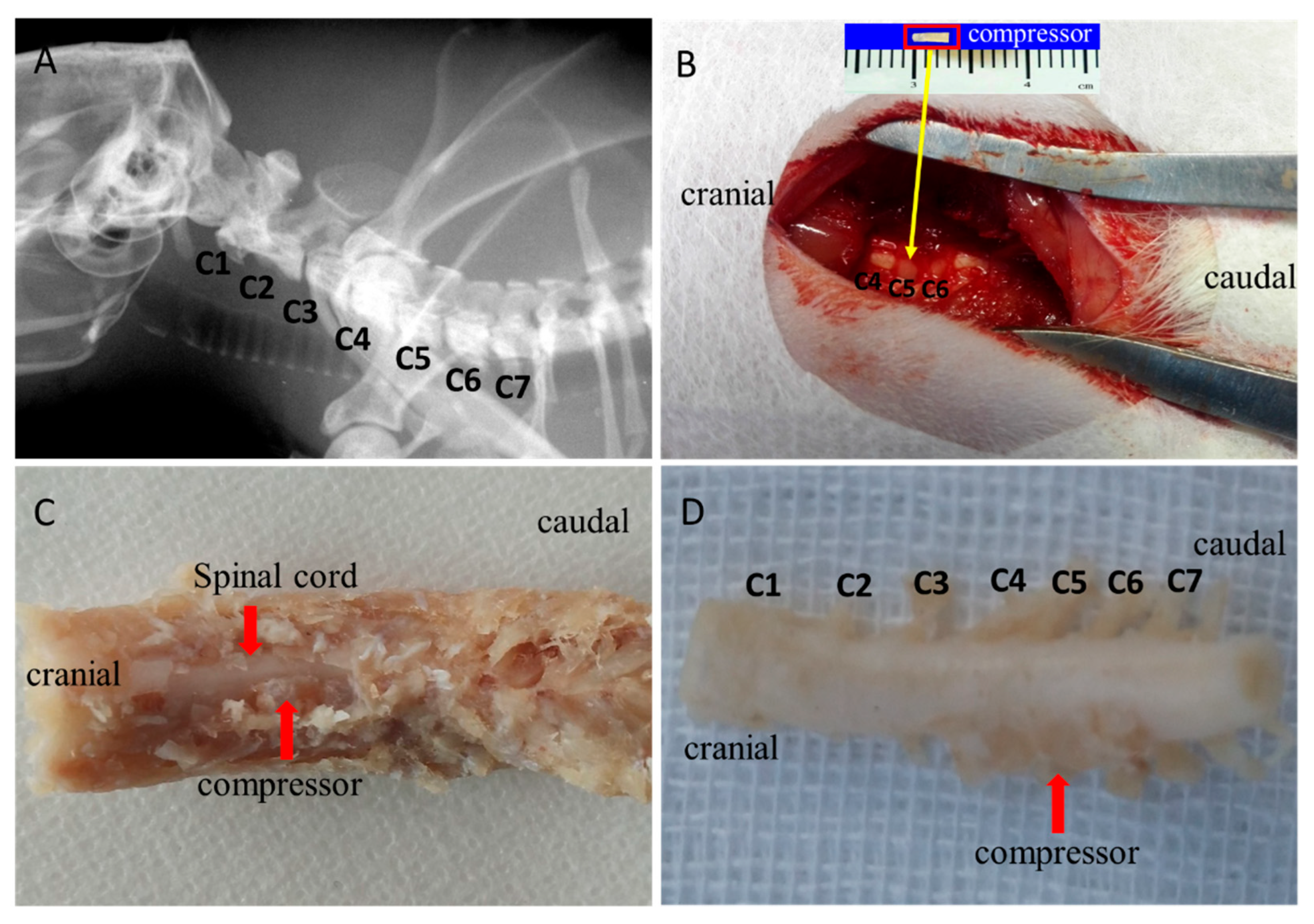
49]. In brief, the animals received general anesthesia with a mixture solution of 10% ketamine and 2% xylazine intraperitoneally. Preoperative location of the cervical spine was identified and marked. After the occipital and nuchal areas were shaved and sterilized, skin incision and thereafter blunt dissection were performed. The surrounding ligamentum flavum between C4 and C5 was exposed under microscopy. A thin polymer sheet was then carefully inserted into the left side of the rat spinal canal at the C5 level (
Figure 1B). After implantation, the incision site was closed by layers. Then, the rats were recovered fully from the surgery on a heating bed and sent back to the cage freely for food and water. The polymer was expanding to reach maximum expansion in 24 h and remain at the maximal volume for 2 months, producing a chronic and persistent course of compression on the cervical spinal cord in the rats.
4.2. Neurological Function Assessment
Locomotor function was evaluated by using the 21-point Basso Beattie Bresnahan (BBB) scoring system [
50]. Two spinal surgeons were invited to evaluate the locomotor function of the rats independently based on the scoring system. Scores were recorded at the timepoints of 1, 3, 7, 14, 21-days, 1-month and 2-month. The average score was calculated to depict the dynamic locomotor function of the modeling rats.
Sensory functional integrity of the cord among the model rats were evaluated by somatosensory evoked potential evaluations (SEP) using an established protocol (Zhang et al., 2009). A constant current stimulation (3.4 Hz square wave, 0.2 ms in duration, 0.3 ms time interval) was transmitted through the simulating electrode into the median nerve at the forelimb of the rat. The cortical SEP was recorded from the skull at Cz–Fz. The signal was amplified 2000 times by a band-pass filter between 10 and 2000 Hz (Zhuhai Yiruikeji Co., Ltd., Zhuhai, China). To obtain a good quality of the SEP signals, a total of 200 SEP responses was averaged for each trial. For synchronization observation of motor sensory function of the rats, SEP raw data were collected at the timepoint, consistent with BBB soring.
4.3. Tissue Preparation and Histopathology Examination
After the experiment, rats were sacrificed by overdose injection of intravenous sodium pentobarbital and perfused with 50 mL heparin saline solution through the ascending aorta thereafter with 300 mL formalin–picric solution (4% formaldehyde, 0.4% picric acid in 0.16 mol/L phosphate buffer, pH = 7.4). The whole cervical spinal cord was carefully harvested and fixed with 4% phosphate buffer liquid in formaldehyde solution for another 72 h. The compressive epicenter and adjacent level (5 mm caudal to the compressive epicenter) of the cord were cut, respectively, and embedded in paraffin. The specimens were continuously sectioned into transverse or sagittal slices of 4 μm thickness using a microtome.
The section specimens were stained with hematoxylin-eosin (H&E) and Luxol Fast blue (LFB, Sigma Chemical Co., St. Louis, MO, USA) staining. All images of the cords were acquired by a microscopic imaging system (FV-1000, Olympus, Japan). H&E staining was used to verify neuronal and axonal degeneration. All large motor neurons with clearly delineated centrally located nuclei and abundant Nissl substance within the perikarya were identified and counted in the ventral horn of the GM at ×10 view among all model rats using Image J 1.47 V. LFB was employed to stain the neurokeratin of myelinated fibers mainly in the WM of the spinal cord. The blue color intensity indicated the content of myelin.
4.4. Immunohistochemistry
After deparaffinating, the sections were treated with a 3% solution of hydrogen peroxide (H2O2) in 95% methanol (MeOH) to block endogenous peroxidase activity. The sections were rinsed in 0.01 M PBS (3 × 5 min). Then, the tissues were treated with 1:100 trypsin solution and 10 mM warm citrate buffer to unmask hidden antigens. The sections were rinsed in 0.01 MPBS solution (3 × 5 min) and the tissue was incubated for 30 min in 1:10 solution of normal goat serum in PBS at 37 °C. The solution was removed and the tissue was incubated in the dilutions of monoclonal Anti-Rat Blood–Brain Barrier antibody (Covance, Cat.No.SMI-71R, 1:1000), Anti-OLIG2 antibody (Bioss, bs-11194R, 1:200), Anti-AIF1/Iba1 antibody (Bioss, bs-1363R, 1:200) and Anti-GFAP antibody (Bioss, bs-0199R, 1:400) at 4 °C overnight. The primary antibody substituted for PBS was set for the negative control. The sections were incubated in the biotinylated secondary antibody, i.e., 1:20 goat anti-mouse IgG (Vector Laboratories, Inc., Cat. No. BP-9200) and goat anti-rabbit IgG (ZSGB-BIO, SP9001) for 1 h at 37 °C, respectively, rinsed in PBS (3 × 5 min) then added to an avidin–biotin–peroxidase complex (Elite® ABC reagent, Cat. No.PK-7100) for 30 min. We used 3,3-diaminobenzidine (DAB) kit (Vector Laboratories, Inc., Cat. No. SK-4100) as visualization chromogen according to the manufacturer’s instructions. The tissues were incubated with DAB substrate for 30 s to 4 min. For quantitative analysis of neuroglial cells and their correlation with microvessels, nuclei of oligodendrocytes, astrocytes and microglial cells were counterstained with Mayer’s hematoxylin. The counterstain recipe can give clear and sharp nucleus staining with little background. After being dehydrated in a gradient of ethanol and cleared with xylene, the sections were cover-slipped using DPX Mountant and dried for microscopic observation.
4.5. Evan Blue Extravasation Measurement
For quantitative and qualitative analysis of the BSCB dysfunction severity, the contents of the extravasating EB and fluorescence were detected. The optical density (OD) value was measured by a spectrophotometer at 620 nm wavelength. The standard curve was drawn and the corresponding linear regression equation was calculated according to the testing result of EB solution at different concentrations (6, 3, 1.5, 0.75, 0.35, 0.175 μg/mL). A 2% EB solution (10 mL/kg; Sigma) was slowly intravenously administered through the vena caudalis. The solution was allowed 24 h for microcirculation and sufficient extravasation into the spinal cord parenchyma in vivo. After satisfactory anesthesia, the rats were transcardially perfused with 500 mL/kg saline. The intact cervical spinal cord was harvested and weighed, then kept in 3 mL methanamide solution for 72 h (37 °C). The cord tissues were centrifuged at 1500 r/min for 15 min. The supernatant was obtained to measure the OD value at 620 nm and the EB concentration can be calculated according to the standard cure. The EB content was calculated based on the following equation:
On the other side, the spinal cord tissue was frozen in liquid nitrogen and then embedded in OCT. Slices at 20 μm were sectioned and visualized using a confocal laser scan microscope system (Germany Leica, TCS SPS II; Olympus OLS-3100).
4.6. Transmission Electron Microscopy Investigation
Rats underwent satisfactory euthanasia with sodium pentobarbital solution. Transcardiac perfusion was not carried out in order to preserve the ultrastructure of spinal cord capillaries. The cervical spines of the rats were harvested and fixed in 4% paraformaldehyde (PFA) in 0.1 M PBS solution (PH 7.0) for 24 h at 4 °C. Subsequently, careful anatomical separation was performed to obtain an integral cervical spinal cord. Sagittal spinal cord fragments (3 mm × 1 mm × 1 mm) from the posterior funiculus of WM and GM were separated and then fixed with 2.5% glutaraldehyde in phosphate buffer (pH7.0) overnight. The cord specimens were washed three times in the PBS for 15 min and then postfixed with 1% OsO4 in PBS for 1h. After dehydration by a graded series of ethanol, specimens were embedded in Epon medium and heated at 70 °C for about 9 h. Finally, the specimen sections at 90 nm were stained by uranyl acetate and alkaline lead citrate, respectively, for 15 min and observed in TEM (JEOL, JEM-1400). Images acquired from imaging system (OLYMPUS DP74) were used to investigate ultrastructural features of neuro-vascular cytohistology.
For quantitative analysis of the ultrastructure of the microvessel and the neuron, as well as the neuroglial cells, micrographs (4008 × 2672 pixels, 24.559 × 16.373 μm, 6.137 nm/pixel) of TEM were acquired and analyzed using Image J 1.47V. The analyzing protocol was based on a previous ultrastructural study [
16].
4.7. Quantitative Analysis of EBA-Immunopositive Microvessel and Neural Cells
On each anatomic region of the whole spinal cord, such as VH and DH of GM, AF, PF and LF of WM, 5 fields of ×20 view (294 × 236 μm2) image were acquired with constant imaging parameter by a microscopic imaging system (FV-1000, Olympus, Japan). For quantitative analysis of blood vessels, the selected regional EBA immunostained vascular profile per filed was outlined by Image J 1.47 V. Then, the RVPN and RVPA per filed were automatically computed. Average RVPN and average RVPA were calculated in different anatomic regions. Percentages of the average RVPN and RVPA were also calculated to explore the distribution of vasculature in the spinal cord.
To quantitatively analyze the neuroglial cells, two practiced pathology technicians were invited to classify and count the number of oligodendrocytes, astrocytes and microglial cells independently on ×20 images. The types of neuroglia cells were primarily distinguished based on morphologic features of the nucleus, which were presented in the EBA immunostaining section. The quantity of neuroglia cells was counted in different anatomic regions. Then, mean numbers of oligodendrocytes, astrocytes and microglial cells were computed by averaging the results from the pathology technicians.
4.8. Statistical Analysis
For intra-group comparison of RVPN, RVPA and neuroglial cell number in different anatomic regions, an independent t-test was performed by using SPSS 16.0 (SPSS Inc., Chicago, IL, USA). For inter-group comparison among control group, compressive epicenter and adjacent level, a one-way analysis of variance (ANOVA) and post hoc test were performed. Matrix Scatter Plot and Linear Correlation analysis were used to explore the relationship between RVPN, RVPA and neuroglia cells. Data are presented as mean values ± SEM and p < 0.05 was considered statistic significant.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









