
Human Cytochrome P450 1, 2, 3 Families as Pharmacogenes with Emphases on Their Antimalarial and Antituberculosis Drugs and Prevalent African Alleles
 , , ,
, , , 
Abstract
:1. Introduction
2. CYP Enzymes
2.1. Human CYP Enzyme Nomenclature
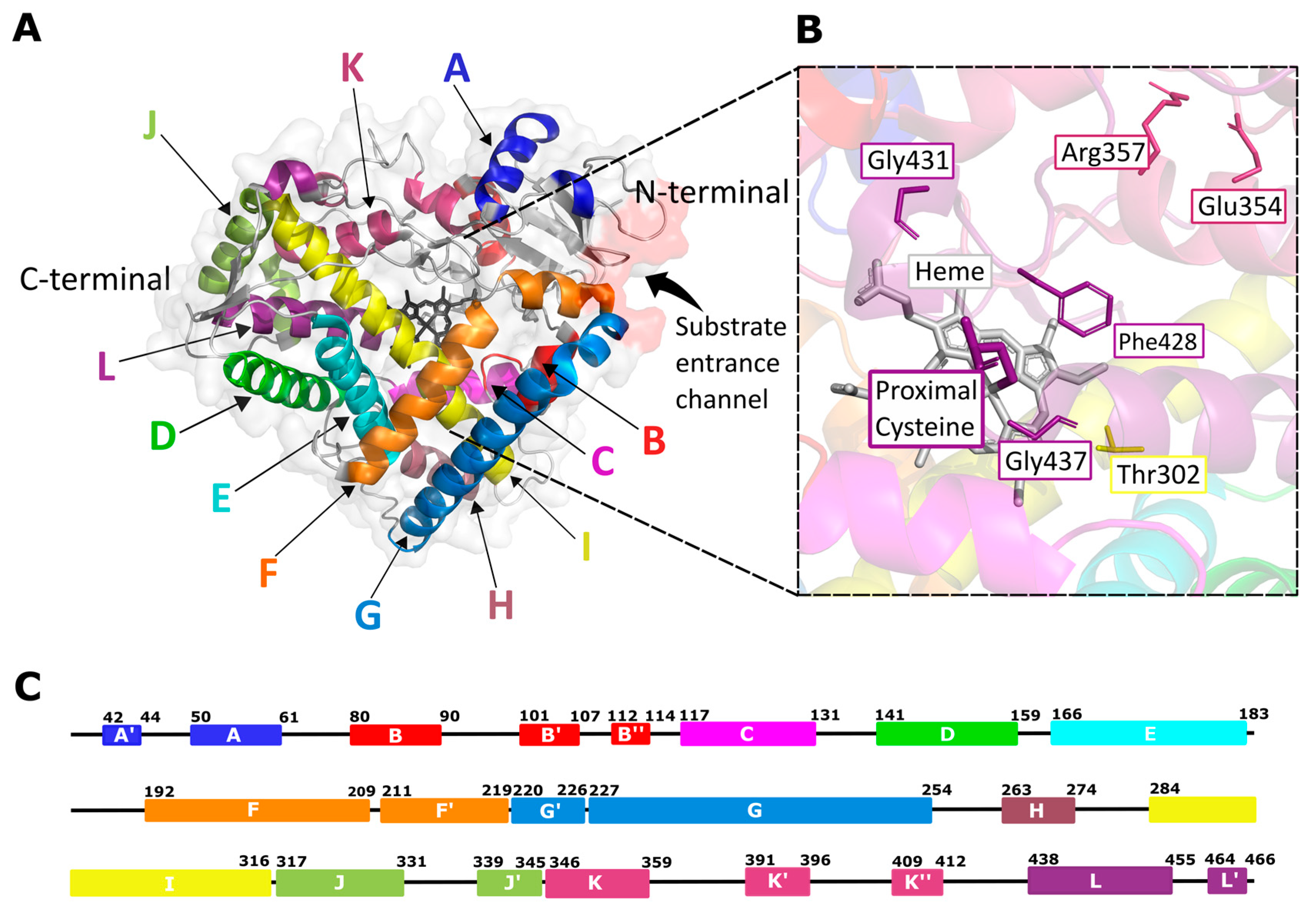
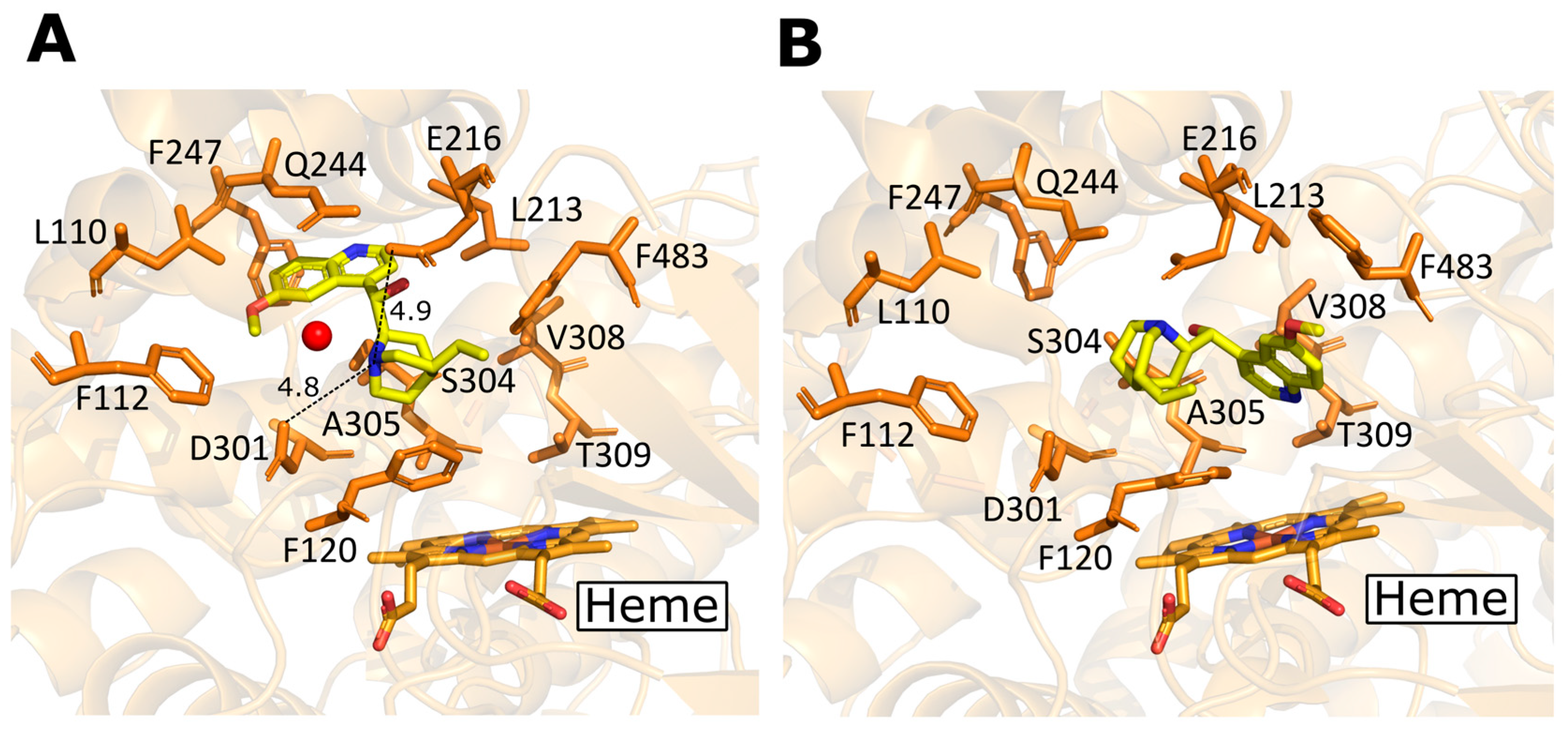
2.2. Human CYP Enzyme Sequence, Structure, and Mechanism of Action

2.3. Existing Structural Information in the Protein Data Bank (PDB)
Antimalarial and Antituberculosis Drugs Complexed with CYP Enzymes
2.4. Structural Differences in Human CYP 1, 2, 3 Enzymes
3. CYP 1, 2, 3 Enzyme Families and Antimalarial Drugs
4. CYP 1, 2, 3 Enzyme Families and Antituberculosis Drugs
5. Inhibition and/or Activation of CYP Enzymes by Drugs, and Drug–Drug Interactions
6. Polymorphism in CYP Enzymes with Specific Focus on Antimalarial and Antituberculosis Drugs
Afrocentric Missense Mutations
7. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ganguly, A. Current Overview of Anti-Tuberculosis Drugs: Metabolism and Toxicities. Mycobact. Dis. 2016, 6, 1000209. [Google Scholar] [CrossRef]
- Liu, X.; Ren, S.; Zhang, J.; Xu, D.; Jiang, F.; Jiang, P.; Feng, J.; Deng, F. The association between cytochrome P450 polymorphisms and anti-tuberculosis drug-induced liver injury: A systematic review and meta-analysis. Ann. Palliat. Med. 2021, 10, 6518–6534. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.-Q. Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef]
- Hassan, R.; Allali, I.; Agamah, F.E.; Elsheikh, S.S.M.; Thomford, N.E.; Dandara, C.; Chimusa, E.R. Drug response in association with pharmacogenomics and pharmacomicrobiomics: Towards a better personalized medicine. Brief. Bioinform. 2020, 22, bbaa292. [Google Scholar] [CrossRef]
- Johnson, J.A. Ethnic Differences in Cardiovascular Drug Response. Circulation 2008, 118, 1383–1393. [Google Scholar] [CrossRef]
- Wilke, R.A.; Lin, D.W.; Roden, D.M.; Watkins, P.B.; Flockhart, D.; Zineh, I.; Giacomini, K.M.; Krauss, R.M. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat. Rev. Drug Discov. 2007, 6, 904–916. [Google Scholar] [CrossRef]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- Schärfe, C.P.I.; Tremmel, R.; Schwab, M.; Kohlbacher, O.; Marks, D.S. Genetic variation in human drug-related genes. Genome Med. 2017, 9, 117. [Google Scholar] [CrossRef]
- Hoehe, M.R.; Kroslak, T. Genetic variation and pharmacogenomics: Concepts, facts, and challenges. Dialog. Clin. Neurosci. 2004, 6, 5–26. [Google Scholar] [CrossRef]
- Weinshilboum, R. Inheritance and Drug Response. N. Engl. J. Med. 2003, 348, 529–537. [Google Scholar] [CrossRef]
- Katara, P.; Yadav, A. Pharmacogenes (PGx-genes): Current understanding and future directions. Gene 2019, 718, 144050. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2021, 29, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z. The Drug Transporter-Metabolism Alliance: Uncovering and Defining the Interplay. Mol. Pharm. 2009, 6, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.C.; Yang, E.H.; Lapetina, D.; Carr, M.S.; Yavorskyy, V.; Hague, J.; Aitchison, K.J. How Can Drug Metabolism and Transporter Genetics Inform Psychotropic Prescribing? Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Petzinger, E.; Geyer, J. Drug transporters in pharmacokinetics. Naunyn. Schmiedeberg. Arch. Pharmacol. 2006, 372, 465–475. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Gaedigk, A.; Ingelman-Sundberg, M.; Miller, N.A.; Leeder, J.S.; Whirl-Carrillo, M.; Klein, T.E.; PharmVar Steering Committee. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 2018, 103, 399–401. [Google Scholar] [CrossRef]
- Preissner, S.C.; Hoffmann, M.F.; Preissner, R.; Dunkel, M.; Gewiess, A.; Preissner, S. Polymorphic Cytochrome P450 Enzymes (CYPs) and Their Role in Personalized Therapy. PLoS ONE 2013, 8, e82562. [Google Scholar] [CrossRef]
- Rajman, I.; Knapp, L.; Morgan, T.; Masimirembwa, C. African Genetic Diversity: Implications for Cytochrome P450-mediated Drug Metabolism and Drug Development. eBioMedicine 2017, 17, 67–74. [Google Scholar] [CrossRef]
- Marwa, K.J.; Schmidt, T.; Sjögren, M.; Minzi, O.M.; Kamugisha, E.; Swedberg, G. Cytochrome P450 single nucleotide polymorphisms in an indigenous Tanzanian population: A concern about the metabolism of artemisinin-based combinations. Malar. J. 2014, 13, 420. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges; World Health Organization: Geneva, Switzerland, 2020; Available online: https://reliefweb.int/attachments/5adfc2b9-0e73-3c8b-97cd-c197b6864385/WMR-2020-v5-double-embargoed.pdf (accessed on 17 November 2022).
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Human drug metabolising cytochrome P450 enzymes: Properties and polymorphisms. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 369, 89–104. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.L.; Lim, C.W.; Sim, W.C.; Toh, L.X.; Leong, K.P. Analysis of Genetic Variation in CYP450 Genes for Clinical Implementation. PLoS ONE 2017, 12, e0169233. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Inui, H. Metabolism of agrochemicals and related environmental chemicals based on cytochrome P450s in mammals and plants. Pest Manag. Sci. 2014, 71, 824–828. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Nebert, D.W.; Adesnik, M.; Coon, M.J.; Estabrook, R.W.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F. The P450 Gene Superfamily: Recommended Nomenclature; Mary Ann Liebert, Inc.: Larchmont, NY, USA, 1987. [Google Scholar]
- Nebert, D.W.; Nelson, D.R.; Adesnik, M.; Coon, M.J.; Estabrook, R.W.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; Kemper, B.; et al. The P450 Superfamily: Updated Listing of All Genes and Recommended Nomenclature for the Chromosomal Loci; Mary Ann Liebert, Inc.: Larchmont, NY, USA, 1989. [Google Scholar]
- Nebert, D.W.; Nelson, D.R.; Coon, M.J.; Estabrook, R.W.; Feyereisen, R.; Fujii-Kuriyama, Y.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; et al. The P450 Superfamily: Update on New Sequences, Gene Mapping, and Recommended Nomenclature; Mary Ann Liebert, Inc.: Larchmont, NY, USA, 1991. [Google Scholar]
- Nelson, D.; Kamataki, T.; Waxman, D.; Guengerich, F.P.; Estabrook, R.W.; Feyereisen, R.; Gonzalez, F.J.; Coon, M.J.; Gunsalus, I.C.; Gotoh, O.; et al. The P450 Superfamily: Update on New Sequences, Gene Mapping, Accession Numbers, Early Trivial Names of Enzymes, and Nomenclature; Mary Ann Liebert, Inc.: Larchmont, NY, USA, 1993. [Google Scholar]
- Nelson, D.R.; Zeldin, D.C.; Hoffman, S.M.; Maltais, L.J.; Wain, H.M.; Nebert, D.W. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 2004, 14, 1–18. [Google Scholar] [CrossRef]
- Daly, A.K.; Brockmoller, J.; Broly, F.; Eichelbaum, M.; Evans, W.; Gonzalez, F.J.; Huang, J.-D.; Idle, J.R.; Ingelman-Sundberg, M.; Ishizaki, T.; et al. Nomenclature for human CYP2D6 alleles. Pharmacogenetics 1996, 6, 193–201. [Google Scholar] [CrossRef]
- Sim, S.C.; Ingelman-Sundberg, M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: A peer-reviewed database of CYP variants and their associated effects. Hum. Genom. 2010, 4, 278–281. [Google Scholar] [CrossRef]
- Sim, S.C.; Ingelman-Sundberg, M. Update on Allele Nomenclature for Human Cytochromes P450 and the Human Cytochrome P450 Allele (CYP-Allele) Nomenclature Database. Methods Mol. Biol. 2013, 987, 251–259. [Google Scholar] [CrossRef]
- Danielson, P.B. The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef] [PubMed]
- Esteves, F.; Rueff, J.; Kranendonk, M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. J. Xenobiotics 2021, 11, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.-T.; Nguyen, N.-L.; Tung, N.; Nguyen, H.; Milhim, M.; Le, T.-T.; Lai, T.-H.; Phan, T.-T.; Bernhardt, R. A Novel Thermostable Cytochrome P450 from Sequence-Based Metagenomics of Binh Chau Hot Spring as a Promising Catalyst for Testosterone Conversion. Catalysts 2020, 10, 1083. [Google Scholar] [CrossRef]
- Sarparast, M.; Dattmore, D.; Alan, J.; Lee, K.S.S. Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients 2020, 12, 3523. [Google Scholar] [CrossRef] [PubMed]
- Machalz, D.; Pach, S.; Bermudez, M.; Bureik, M.; Wolber, G. Structural insights into understudied human cytochrome P450 enzymes. Drug Discov. Today 2021, 26, 2456–2464. [Google Scholar] [CrossRef]
- Mustafa, G.; Nandekar, P.P.; Mukherjee, G.; Bruce, N.J.; Wade, R.C. The Effect of Force-Field Parameters on Cytochrome P450-Membrane Interactions: Structure and Dynamics. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Poulos, T.L.; Johnson, E.F. Structures of Cytochrome P450 Enzymes. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: Cham, Switzerland, 2015; pp. 3–32. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Textbook of Biochemistry with Clinical Correlations, 7th Edition|Wiley. In: Wiley.com. Available online: https://www.wiley.com/en-us/Textbook+of+Biochemistry+with+Clinical+Correlations%2C+7th+Edition-p-9780470281734 (accessed on 26 August 2022).
- Midlik, A.; Navrátilová, V.; Moturu, T.R.; Koča, J.; Svobodová, R.; Berka, K. Uncovering of cytochrome P450 anatomy by SecStrAnnotator. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinković, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Munro, A.W.; Girvan, H.M.; Mason, A.E.; Dunford, A.J.; McLean, K.J. What makes a P450 tick? Trends Biochem. Sci. 2013, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Fishelovitch, D.; Shaik, S.; Wolfson, H.J.; Nussinov, R. Theoretical Characterization of Substrate Access/Exit Channels in the Human Cytochrome P450 3A4 Enzyme: Involvement of Phenylalanine Residues in the Gating Mechanism. J. Phys. Chem. B 2009, 113, 13018–13025. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta (BBA) 2007, 1770, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Dubey, K.D.; Shaik, S. Cytochrome P450—The Wonderful Nanomachine Revealed through Dynamic Simulations of the Catalytic Cycle. Acc. Chem. Res. 2019, 52, 389–399. [Google Scholar] [CrossRef]
- Hu, Q.; Hartmann, R.W. The Renaissance of CYP17 Inhibitors for the Treatment of Prostate Cancer. In Cancer Drug Design and Discovery; Elsevier: Amsterdam, The Netherlands, 2014; pp. 319–356. [Google Scholar]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Coleman, T.; Stok, J.E.; Podgorski, M.N.; Bruning, J.B.; De Voss, J.J.; Bell, S.G. Structural insights into the role of the acid-alcohol pair of residues required for dioxygen activation in cytochrome P450 enzymes. JBIC J. Biol. Inorg. Chem. 2020, 25, 583–596. [Google Scholar] [CrossRef]
- Wang, B.; Li, C.; Dubey, K.D.; Shaik, S. Quantum Mechanical/Molecular Mechanical Calculated Reactivity Networks Reveal How Cytochrome P450cam and Its T252A Mutant Select Their Oxidation Pathways. J. Am. Chem. Soc. 2015, 137, 7379–7390. [Google Scholar] [CrossRef]
- Wu, J.; Guan, X.; Dai, Z.; He, R.; Ding, X.; Yang, L.; Ge, G. Molecular probes for human cytochrome P450 enzymes: Recent progress and future perspectives. Coord. Chem. Rev. 2020, 427, 213600. [Google Scholar] [CrossRef]
- Ueyama, N.; Nishikawa, N.; Yamada, Y.; Okamura, T.-A.; Nakamura, A. Cytochrome P-450 Model (Porphinato)(thiolato)iron(III) Complexes with Single and Double NH···S Hydrogen Bonds at the Thiolate Site. J. Am. Chem. Soc. 1996, 118, 12826–12827. [Google Scholar] [CrossRef]
- Ueyama, N.; Terakawa, T.; Nakata, M.; Nakamura, A. Positive shift of redox potential of [Fe4S4(Z-cys-Gly-Ala-OMe)4]2- in dichloromethane. J. Am. Chem. Soc. 1983, 105, 7098–7102. [Google Scholar] [CrossRef]
- Matsunaga, I.; Sumimoto, T.; Ueda, A.; Kusunose, E.; Ichihara, K. Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis: Substrate structure required for α-hydroxylation. Lipids 2000, 35, 365–371. [Google Scholar] [CrossRef]
- Lee, D.-S.; Yamada, A.; Sugimoto, H.; Matsunaga, I.; Ogura, H.; Ichihara, K.; Adachi, S.-I.; Park, S.-Y.; Shiro, Y. Substrate Recognition and Molecular Mechanism of Fatty Acid Hydroxylation by Cytochrome P450 from Bacillus subtilis. J. Biol. Chem. 2003, 278, 9761–9767. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular Mechanism of Phase I and Phase II Drug-Metabolizing Enzymes: Implications for Detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [CrossRef] [PubMed]
- Vasav, A.P.; Barvkar, V.T. Phylogenomic analysis of cytochrome P450 multigene family and their differential expression analysis in Solanum lycopersicum L. suggested tissue specific promoters. BMC Genom. 2019, 20, 116. [Google Scholar] [CrossRef] [PubMed]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends Biotechnol. 2012, 30, 26–36. [Google Scholar] [CrossRef]
- Van Bogaert, I.N.A.; Groeneboer, S.; Saerens, K.; Soetaert, W. The role of cytochrome P450 monooxygenases in microbial fatty acid metabolism. FEBS J. 2010, 278, 206–221. [Google Scholar] [CrossRef]
- Henry, H.L. Cytochrome P450 Structure, Mechanism, and Biochemistry, 2nd ed.; Edited by Paul R. Oritz de Montellano (University of California, San Francisco). Plenum: New York. 1995. xi + 631 pp. $125.00. ISBN 0-306-45141-7. J. Am. Chem. Soc. 1996, 118, 10945. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.H.; Sono, M. Cytochrome P-450 and chloroperoxidase: Thiolate-ligated heme enzymes. Spectroscopic determination of their active-site structures and mechanistic implications of thiolate ligation. Chem. Rev. 1987, 87, 1255–1276. [Google Scholar] [CrossRef]
- de Montellano, P.R.O.; De Voss, J.J. Oxidizing species in the mechanism of cytochrome P450. Nat. Prod. Rep. 2002, 19, 477–493. [Google Scholar] [CrossRef]
- Hoa, G.H.B.; McLean, M.; Sligar, S.G. High pressure, a tool for exploring heme protein active sites. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 2002, 1595, 297–308. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Macdonald, T.L. Mechanisms of cytochrome P-450 catalysis. FASEB J. 1990, 4, 2453–2459. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations. Chem. Rev. 2009, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, O. An odyssey with oxygen. Biochem. Biophys. Res. Commun. 2005, 338, 2–6. [Google Scholar] [CrossRef]
- Waterman, M.R. Professor Howard Mason and oxygen activation. Biochem. Biophys. Res. Commun. 2005, 338, 7–11. [Google Scholar] [CrossRef]
- Mowat, C.G.; Gazur, B.; Campbell, L.P.; Chapman, S.K. Flavin-containing heme enzymes. Arch. Biochem. Biophys. 2009, 493, 37–52. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of cytochrome P450 substrate oxidation: Mini Review. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef]
- Zheng, J.; Altun, A.; Thiel, W. Common system setup for the entire catalytic cycle of cytochrome P450cam in quantum mechanical/molecular mechanical studies. J. Comput. Chem. 2007, 28, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Sligar, S.G.; Gunsalus, I.C. A thermodynamic model of regulation: Modulation of redox equilibria in camphor monoxygenase. Proc. Natl. Acad. Sci. USA 1976, 73, 1078–1082. [Google Scholar] [CrossRef]
- Govindaraj, S.; Poulos, T.L. The Domain Architecture of Cytochrome P450BM-3. J. Biol. Chem. 1997, 272, 7915–7921. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.J.; Rock, D.A.; Jones, J.P. Evidence for Two Different Active Oxygen Species in Cytochrome P450 BM3 Mediated Sulfoxidation and N-Dealkylation Reactions. J. Am. Chem. Soc. 2002, 124, 9724–9725. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cojocaru, V.; Wade, R.C. Conformational diversity and ligand tunnels of mammalian cytochrome P450s. Biotechnol. Appl. Biochem. 2013, 60, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Stout, C.; Zhang, Q.; Johnson, E.F. Contributions of Ionic Interactions and Protein Dynamics to Cytochrome P450 2D6 (CYP2D6) Substrate and Inhibitor Binding. J. Biol. Chem. 2015, 290, 5092–5104. [Google Scholar] [CrossRef]
- Dubey, K.D.; Shaik, S. Choreography of the Reductase and P450BM3 Domains toward Electron Transfer Is Instigated by the Substrate. J. Am. Chem. Soc. 2018, 140, 683–690. [Google Scholar] [CrossRef]
- Dubey, K.D.; Wang, B.; Vajpai, M.; Shaik, S. MD simulations and QM/MM calculations show that single-site mutations of cytochrome P450BM3 alter the active site’s complexity and the chemoselectivity of oxidation without changing the active species. Chem. Sci. 2017, 8, 5335–5344. [Google Scholar] [CrossRef]
- Dubey, K.D.; Wang, B.; Shaik, S. Molecular Dynamics and QM/MM Calculations Predict the Substrate-Induced Gating of Cytochrome P450 BM3 and the Regio- and Stereoselectivity of Fatty Acid Hydroxylation. J. Am. Chem. Soc. 2016, 138, 837–845. [Google Scholar] [CrossRef]
- Ramanan, R.; Dubey, K.D.; Wang, B.; Mandal, D.; Shaik, S. Emergence of Function in P450-Proteins: A Combined Quantum Mechanical/Molecular Mechanical and Molecular Dynamics Study of the Reactive Species in the H2O2-Dependent Cytochrome P450SPα and Its Regio and Enantioselective Hydroxylation of Fatty Acids. J. Am. Chem. Soc. 2016, 138, 6786–6797. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Savas, U.; Stout, C.D.; Johnson, E.F. Structural Characterization of the Complex between α-Naphthoflavone and Human Cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. [Google Scholar] [CrossRef]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand Access Channels in Cytochrome P450 Enzymes: A Review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human Cytochrome P450 1A1 Structure and Utility in Understanding Drug and Xenobiotic Metabolism*. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef]
- McLaughlin, L.A.; Paine, M.J.I.; Kemp, C.A.; Maréchal, J.-D.; Flanagan, J.U.; Ward, C.J.; Sutcliffe, M.; Roberts, G.; Wolf, C.R. Why Is Quinidine an Inhibitor of Cytochrome P450 2D6? J. Biol. Chem. 2005, 280, 38617–38624. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, J.M.; Walker, G.S.; Wienkers, L.C. Inhibition of Cytochrome P450 2D6: Structure−Activity Studies Using a Series of Quinidine and Quinine Analogues. Chem. Res. Toxicol. 2003, 16, 450–459. [Google Scholar] [CrossRef]
- Otton, S.; Inaba, T.; Kalow, W. Competitive inhibition of sparteine oxidation in human liver by β-adrenoceptor antagonists and other cardiovascular drugs. Life Sci. 1984, 34, 73–80. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Mahonen, N.; Raunio, H.; Rautio, J. Cytochrome P450-Activated Prodrugs: Targeted Drug Delivery. Curr. Med. Chem. 2008, 15, 2346–2365. [Google Scholar] [CrossRef]
- Yano, J.K.; Hsu, M.-H.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 2005, 12, 822–823. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Vinković, D.M.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef]
- Wester, M.R.; Yano, J.K.; Schoch, G.A.; Yang, C.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Cytochrome P450 2C9 Complexed with Flurbiprofen at 2.0-Å Resolution. J. Biol. Chem. 2004, 279, 35630–35637. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Wu, B.; Chow, D.; Hu, M. Substrate selectivity of drug-metabolizing cytochrome P450s predicted from crystal structures and in silico modeling. Drug Metab. Rev. 2012, 44, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, P.R.; Meneely, K.M.; Scott, E.E. Structures of Human Cytochrome P-450 2E1. J. Biol. Chem. 2008, 283, 33698–33707. [Google Scholar] [CrossRef]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by x-ray crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05-Å Resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Ekroos, M.; Sjögren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Interaction of human cytochrome P4503A4 with ritonavir analogs. Arch. Biochem. Biophys. 2012, 520, 108–116. [Google Scholar] [CrossRef]
- Hsu, M.-H.; Savas, U.; Johnson, E.F. The X-Ray Crystal Structure of the Human Mono-Oxygenase Cytochrome P450 3A5-Ritonavir Complex Reveals Active Site Differences between P450s 3A4 and 3A5. Mol. Pharmacol. 2017, 93, 14–24. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guidelines for Malaria, 3 June 2022; World Health Organization: Geneva, Switzerland, 2022; (WHO/UCN/GMP/2022.01 Rev.2); Available online: https://apps.who.int/iris/rest/bitstreams/1427681/retrieve (accessed on 17 November 2022).
- AlKadi, H.O. Antimalarial Drug Toxicity: A Review. Chemotherapy 2007, 53, 385–391. [Google Scholar] [CrossRef]
- Taylor, W.R.J.; White, N.J. Antimalarial Drug Toxicity. Drug Saf. 2004, 27, 25–61. [Google Scholar] [CrossRef] [PubMed]
- Kerb, R.; Fux, R.; Mörike, K.; Kremsner, P.G.; Gil, J.P.; Gleiter, C.H.; Schwab, M. Pharmacogenetics of antimalarial drugs: Effect on metabolism and transport. Lancet Infect. Dis. 2009, 9, 760–774. [Google Scholar] [CrossRef]
- Khoo, S.; Back, D.; Winstanley, P. The potential for interactions between antimalarial and antiretroviral drugs. Aids 2005, 19, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, S.P.R.A.F.P. Metabolism and Interactions of Chloroquine and Hydroxychloroquine with Human Cytochrome P450 Enzymes and Drug Transporters. Curr. Drug Metab. 2020, 21, 1127–1135. [Google Scholar] [CrossRef]
- Hodel, E.M.S.; Csajka, C.; Ariey, F.; Guidi, M.; Kabanywanyi, A.M.; Duong, S.; Decosterd, L.A.; Olliaro, P.; Beck, H.-P.; Genton, B. Effect of Single Nucleotide Polymorphisms in Cytochrome P450 Isoenzyme and N-Acetyltransferase 2 Genes on the Metabolism of Artemisinin-Based Combination Therapies in Malaria Patients from Cambodia and Tanzania. Antimicrob. Agents Chemother. 2013, 57, 950–958. [Google Scholar] [CrossRef]
- Giao, P.; De Vries, P.J. Pharmacokinetic Interactions of Antimalarial Agents. Clin. Pharmacokinet. 2001, 40, 343–373. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Q.; Björkman, A.; Andersson, T.B.; Ridderström, M.; Masimirembwa, C.M. Amodiaquine Clearance and Its Metabolism to N-Desethylamodiaquine Is Mediated by CYP2C8: A New High Affinity and Turnover Enzyme-Specific Probe Substrate. Experiment 2002, 300, 399–407. [Google Scholar] [CrossRef]
- Zuidema, J.; Hilbers-Modderman, E.S.M.; Merkus, F.W.H.M. Clinical Pharmacokinetics of Dapsone. Clin. Pharmacokinet. 1986, 11, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Zhu, F.; Li, X.; Yang, A.; Xing, J. Auto-induction of phase I and phase II metabolism of artemisinin in healthy Chinese subjects after oral administration of a new artemisinin-piperaquine fixed combination. Malar. J. 2014, 13, 214. [Google Scholar] [CrossRef]
- Ganesan, S.; Sahu, R.; A Walker, L.; Tekwani, B.L. Cytochrome P450-dependent toxicity of dapsone in human erythrocytes. J. Appl. Toxicol. 2009, 30, 271–275. [Google Scholar] [CrossRef]
- Grace, J.M.; Skanchy, D.J.; Aguilar, A.J. Metabolism of artelinic acid to dihydroqinghaosu by human liver cytochrome P4503A. Xenobiotica 1999, 29, 703–717. [Google Scholar] [CrossRef]
- Mehlotra, R.K.; Henry-Halldin, C.N.; A Zimmerman, P. Application of pharmacogenomics to malaria: A holistic approach for successful chemotherapy. Pharmacogenomics 2009, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, G.; Carpenter, P.; Souppart, C.; Schmidli, H.; McClean, M.; Stypinski, D. Pharmacokinetics and electrocardiographic pharmacodynamics of artemether-lumefantrine (Riamet®) with concomitant administration of ketoconazole in healthy subjects. Br. J. Clin. Pharmacol. 2002, 54, 485–492. [Google Scholar] [CrossRef]
- Van Agtmael, M.A.; Gupta, V.; Van Der Wösten, T.H.; Rutten, J.-P.B.; Van Boxtel, C.J. Grapefruit juice increases the bioavailability of artemether. Eur. J. Clin. Pharmacol. 1999, 55, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Mansor, S.M.; Sit, N.W.; Grace, J.; Li, Q.; Olliaro, P. Pharmacokinetics of Artemisinin-Type Compounds. Clin. Pharmacokinet. 2000, 39, 255–270. [Google Scholar] [CrossRef]
- Svensson, U.S.; Ashton, M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br. J. Clin. Pharmacol. 1999, 48, 528–535. [Google Scholar] [CrossRef]
- Woodrow, C.J.; Haynes, R.K.; Krishna, S. Artemisinins. Postgrad. Med. J. 2005, 81, 71–78. [Google Scholar] [CrossRef]
- Yusof, W.; Hua, G.S. Gene, ethnic and gender influences predisposition of adverse drug reactions to artesunate among Malaysians. Toxicol. Mech. Methods 2011, 22, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Sortica, V.A.; Lindenau, J.D.; Cunha, M.G.; Ohnishi, M.D.; Ventura, A.M.R.; Ribeiro-Dos-Santos, K.; Santos, S.E.; Guimarães, L.S.; Hutz, M.H. The effect of SNPs in CYP450 in chloroquine/primaquine Plasmodium vivax malaria treatment. Pharmacogenomics 2016, 17, 1903–1911. [Google Scholar] [CrossRef]
- Projean, D.; Baune, B.; Farinotti, R.; Flinois, J.-P.; Beaune, P.; Taburet, A.-M.; Ducharme, J. In vitro metabolism of chloroquine: Identification of cyp2c8, cyp3a4, and cyp2d6 as the main isoforms catalyzing n-desethylchloroquine formation. Drug Metab. Dispos. 2003, 31, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Winte, H.R.; Wang, Y.; Unadkat, J.D. CYP2C8/9 Mediate Dapsone N-Hydroxylation at Clinical Concentrations of Dapsone. Drug Metab. Dispos. 2000, 28, 865–868. [Google Scholar]
- May, D.G.; Porter, J.; Wilkinson, G.R.; A Branch, R. Frequency distribution of dapsone N-hydroxylase, a putative probe for P4503A4 activity, in a white population. Clin. Pharmacol. Ther. 1994, 55, 492–500. [Google Scholar] [CrossRef]
- A Ward, S.; Sevene, E.J.; Hastings, I.M.; Nosten, F.; McGready, R. Antimalarial drugs and pregnancy: Safety, pharmacokinetics, and pharmacovigilance. Lancet Infect. Dis. 2007, 7, 136–144. [Google Scholar] [CrossRef]
- Pernaute-Lau, L.; Camara, M.; de Sousa, T.N.; Morris, U.; Ferreira, M.U.; Gil, J.P. An update on pharmacogenetic factors influencing the metabolism and toxicity of artemisinin-based combination therapy in the treatment of malaria. Expert Opin. Drug Metab. Toxicol. 2022, 18, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, F.; de Sousa, G.; Burcham, P.; Duchêne, P.; Rahmani, R. Role of cytochrome P450 3A in the metabolism of mefloquine in human and animal hepatocytes. Life Sci. 2000, 66, 2193–2212. [Google Scholar] [CrossRef] [PubMed]
- Na Bangchang, K.; Karbwang, J.; Back, D.J. Mefloquine metabolism by human liver microsomes: Effect of other antimalarial drugs. Biochem. Pharmacol. 1992, 43, 1957–1961. [Google Scholar] [CrossRef]
- Avula, B.; Tekwani, B.L.; Chaurasiya, N.D.; Fasinu, P.; Nanayakkara, N.P.D.; Herath, H.M.T.B.; Wang, Y.-H.; Bae, J.-Y.; Khan, S.I.; ElSohly, M.A.; et al. Metabolism of primaquine in normal human volunteers: Investigation of phase I and phase II metabolites from plasma and urine using ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Malar. J. 2018, 17, 294. [Google Scholar] [CrossRef]
- Somogyi, A.A.; Reinhard, H.A.; Bochner, F. Pharmacokinetic evaluation of proguanil: A probe phenotyping drug for the mephenytoin hydroxylase polymorphism. Br. J. Clin. Pharmacol. 1996, 41, 175–179. [Google Scholar] [CrossRef]
- Zhang, H.; Coville, P.F.; Walker, R.J.; Miners, J.O.; Birkett, D.J.; Wanwimolruk, S. Evidence for involvement of human CYP3A in the 3-hydroxylation of quinine. Br. J. Clin. Pharmacol. 1997, 43, 245–252. [Google Scholar] [CrossRef]
- Nielsen, T.L.; Rasmussen, B.B.; Flinois, J.P.; Beaune, P.; Brosen, K. In Vitro Metabolism of Quinidine: The (3S)-3-Hydroxylation of Quinidine Is a Specific Marker Reaction for Cytochrome P-4503A4 Activity in Human Liver Microsomes. J. Pharmacol. Exp. Ther. 1999, 289, 31–37. [Google Scholar]
- Horsburgh, C.R.; Barry, C.E.; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Calcagno, A.; Bonora, S. Pharmacokinetics and pharmacogenetics of anti-tubercular drugs: A tool for treatment optimization? Expert Opin. Drug Metab. Toxicol. 2017, 14, 59–82. [Google Scholar] [CrossRef]
- Barozi, V.; Musyoka, T.M.; Sheik Amamuddy, O.; Tastan Bishop, Ö. Deciphering Isoniazid Drug Resistance Mechanisms on Dimeric Mycobacterium tuberculosis KatG via Post-molecular Dynamics Analyses Including Combined Dynamic Residue Network Metrics. ACS Omega 2022, 7, 13313–13332. [Google Scholar] [CrossRef]
- Nath, H.; Ryoo, S. First– and Second–Line Drugs and Drug Resistance. Tuberculosis 2013. [Google Scholar] [CrossRef]
- World Health Organisation. Rapid Communication: Key Changes to the Treatment of Drug-Resistant Tuberculosis. 2022. Available online: https://apps.who.int/iris/handle/10665/353743 (accessed on 17 November 2022).
- Sutezolid. In: TB Alliance. Available online: https://www.tballiance.org/portfolio/trial/12018 (accessed on 25 November 2022).
- Zumla, A.; Rao, M.; Wallis, R.S.; E Kaufmann, S.H.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-directed therapies for infectious diseases: Current status, recent progress, and future prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef] [PubMed]
- Magis-Escurra, C.; Boogaard, J.V.D.; Ijdema, D.; Boeree, M.; Aarnoutse, R. Therapeutic drug monitoring in the treatment of tuberculosis patients. Pulm. Pharmacol. Ther. 2012, 25, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Babalik, A.; Mannix, S.; Francis, D.; Menzies, D. Therapeutic Drug Monitoring in the Treatment of Active Tuberculosis. Can. Respir. J. 2011, 18, 225–229. [Google Scholar] [CrossRef]
- Tostmann, A.; Boeree, M.J.; Aarnoutse, R.E.; De Lange, W.C.; van der Ven, A.J.; Dekhuijzen, R. Antituberculosis drug-induced hepatotoxicity: Concise up-to-date review. J. Gastroenterol. Hepatol. 2008, 23, 192–202. [Google Scholar] [CrossRef]
- Shao, Q.; Mao, X.; Zhou, Z.; Huai, C.; Li, Z. Research Progress of Pharmacogenomics in Drug-Induced Liver Injury. Front. Pharmacol. 2021, 12, 735260. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Liu, K.; Li, F.; Lu, J.; Liu, S.; Dorko, K.; Xie, W.; Ma, X. Bedaquiline Metabolism: Enzymes and Novel Metabolites. Drug Metab. Dispos. 2014, 42, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Cuyckens, F.; Balcaen, L.I.L.; De Wolf, K.; De Samber, B.; Van Looveren, C.; Hurkmans, R.; Vanhaecke, F. Use of the bromine isotope ratio in HPLC-ICP-MS and HPLC-ESI-MS analysis of a new drug in development. Anal. Bioanal. Chem. 2008, 390, 1717–1729. [Google Scholar] [CrossRef]
- Meermann, B.; Bockx, M.; Laenen, A.; Van Looveren, C.; Cuyckens, F.; Vanhaecke, F. Speciation analysis of bromine-containing drug metabolites in feces samples from a human in vivo study by means of HPLC/ICP-MS combined with on-line isotope dilution. Anal. Bioanal. Chem. 2011, 402, 439–448. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorganic Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In Vitro Antimycobacterial Spectrum of a Diarylquinoline ATP Synthase Inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef]
- Obach, R.S. Linezolid Metabolism Is Catalyzed by Cytochrome P450 2J2, 4F2, and 1B1. Drug Metab. Dispos. 2022, 50, 413–421. [Google Scholar] [CrossRef]
- Deltyba INN-Delamanid, Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/deltyba-epar-product-information_en.pdf (accessed on 28 November 2022).
- Ericsson, T.; Sundell, J.; Torkelsson, A.; Hoffmann, K.-J.; Ashton, M. Effects of artemisinin antimalarials on Cytochrome P450 enzymes in vitro using recombinant enzymes and human liver microsomes: Potential implications for combination therapies. Xenobiotica 2013, 44, 615–626. [Google Scholar] [CrossRef]
- Nishimura, Y.; Kurata, N.; Sakurai, E.; Yasuhara, H. Inhibitory Effect of Antituberculosis Drugs on Human Cytochrome P450-Mediated Activities. J. Pharmacol. Sci. 2004, 96, 293–300. [Google Scholar] [CrossRef]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. Bayl. Univ. Med. Cent. Proc. 2000, 13, 421–423. [Google Scholar] [CrossRef]
- Shackleford, D.M.; Chiu, F.C.K.; Katneni, K.; Blundell, S.; McLaren, J.; Wang, X.; Zhou, L.; Sriraghavan, K.; Alker, A.M.; Hunziker, D.; et al. Cytochrome P450-Mediated Metabolism and CYP Inhibition for the Synthetic Peroxide Antimalarial OZ439. ACS Infect. Dis. 2021, 7, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Kirby, B.J.; Whittington, D.; Wan, Y.; Goodlett, D.R. Evaluation of P450 Inhibition and Induction by Artemisinin Antimalarials in Human Liver Microsomes and Primary Human Hepatocytes. Drug Metab. Dispos. 2012, 40, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Asimus, S.; Elsherbiny, D.; Hai, T.N.; Jansson, B.; Huong, N.V.; Petzold, M.G.; Simonsson, U.; Ashton, M. Artemisinin antimalarials moderately affect cytochrome P450 enzyme activity in healthy subjects. Fundam. Clin. Pharmacol. 2007, 21, 307–316. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Bi, H.-C.; Xie, Z.-Y.; Zuo, Z.; Li, J.-K.; Li, X.; Zhao, L.-Z.; Chen, X.; Huang, M. Rapid determination of six metabolites from multiple cytochrome P450 probe substrates in human liver microsome by liquid chromatography/mass spectrometry: Application to high-throughput inhibition screening of terpenoids. Rapid Commun. Mass Spectrom. 2007, 21, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Bapiro, T.E.; Egnell, A.-C.; Hasler, J.A.; Masimirembwa, C.M. Application of higher throughput screening (HTS) inhibition assays to evaluate the interaction of antiparasitic drugs with cytochrome P450s. Drug Metab. Dispos. 2001, 29, 30–35. [Google Scholar]
- Bapiro, T.E.; Sayi, J.; Hasler, J.A.; Jande, M.; Rimoy, G.; Masselle, A.; Masimirembwa, C.M. Artemisinin and thiabendazole are potent inhibitors of cytochrome P450 1A2 (CYP1A2) activity in humans. Eur. J. Clin. Pharmacol. 2005, 61, 755–761. [Google Scholar] [CrossRef]
- Ericsson, T.; Masimirembwa, C.; Abelo, A.; Ashton, M. The evaluation of CYP2B6 inhibition by artemisinin antimalarials in recombinant enzymes and human liver microsomes. Drug Metab. Lett. 2013, 6, 247–257. [Google Scholar] [CrossRef]
- Burk, O.; Arnold, K.A.; Nussler, A.K.; Schaeffeler, E.; Efimova, E.; Avery, B.A.; Avery, M.A.; Fromm, M.F.; Eichelbaum, M. Antimalarial Artemisinin Drugs Induce Cytochrome P450 and MDR1 Expression by Activation of Xenosensors Pregnane X Receptor and Constitutive Androstane Receptor. Mol. Pharmacol. 2005, 67, 1954–1965. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. Pxr, car and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef]
- Haddad, A.; Davis, M.; Lagman, R. The pharmacological importance of cytochrome CYP3A4 in the palliation of symptoms: Review and recommendations for avoiding adverse drug interactions. Support. Care Cancer 2006, 15, 251–257. [Google Scholar] [CrossRef]
- Ademisoye, A.A.; Soyinka, J.O.; Olawoye, S.O.; Igbinoba, S.I.; Olowookere, S.A.; Ademisoye, A.T.; Onyeji, C. Induction of Amodiaquine Metabolism by Rifampicin Following Concurrent Administration in Healthy Volunteers. J. Explor. Res. Pharmacol. 2018, 3, 71–77. [Google Scholar] [CrossRef]
- Sahasrabudhe, V.; Zhu, T.; Vaz, A.; Tse, S. Drug Metabolism and Drug Interactions: Potential Application to Antituberculosis Drugs. J. Infect. Dis. 2015, 211, S107–S114. [Google Scholar] [CrossRef] [PubMed]
- Ramappa, V.; Aithal, G.P. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J. Clin. Exp. Hepatol. 2013, 3, 37–49. [Google Scholar] [CrossRef]
- Cho, H.-J.; Koh, W.-J.; Ryu, Y.-J.; Ki, C.-S.; Nam, M.-H.; Kim, J.-W.; Lee, S.-Y. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis 2007, 87, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Sim, E.; Abuhammad, A.; Ryan, A. Arylamine N-acetyltransferases: From drug metabolism and pharmacogenetics to drug discovery. Br. J. Pharmacol. 2014, 171, 2705–2725. [Google Scholar] [CrossRef]
- Sandy, J.; Holton, S.; Fullam, E.; Sim, E.; Noble, M. Binding of the anti-tubercular drug isoniazid to the arylamine N-acetyltransferase protein from Mycobacterium smegmatis. Protein Sci. 2005, 14, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Boukouvala, S.; McDonagh, E.M.; Shuldiner, S.R.; Laurieri, N.; Thorn, C.F.; Altman, R.; Klein, T.E. PharmGKB summary. Pharmacogenetics Genom. 2016, 26, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Timbrell, J.A. Factors affecting the metabolism of [14C] acetylhydrazine in rats. Drug Metab. Dispos. 1978, 6, 561–566. [Google Scholar] [PubMed]
- Timbrell, J.A. Young Scientists Award Lecture 1978: Studies on the Role of Acetylhydrazine in Isoniazid Hepatotoxicity. Arch. Toxicol. 1979, 2, 1–8. [Google Scholar] [CrossRef]
- Meitei, H.N.; Pandey, A.; Haobam, R. Polymorphisms in drug metabolism genes as a risk factor for first-line anti-tuberculosis drug-induced liver injury. Mol. Biol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Rodulfo, A.; Carrillo-Tripp, M.; Yeverino-Gutierrez, M.L.; Peñuelas-Urquides, K.; González-Escalante, L.A.; de León, M.B.; Silva-Ramirez, B. NAT2 polymorphisms associated with the development of hepatotoxicity after first-line tuberculosis treatment in Mexican patients: From genotype to molecular structure characterization. Clin. Chim. Acta 2021, 519, 153–162. [Google Scholar] [CrossRef]
- Wang, P.-Y.; Xie, S.-Y.; Hao, Q.; Zhang, C.; Jiang, B.-F. NAT2 polymorphisms and susceptibility to anti-tuberculosis drug-induced liver injury: A meta-analysis [Review article]. Int. J. Tuberc. Lung Dis. 2012, 16, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Day, C.P. Genetic association studies in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Krausz, K.W.; Li, F.; Ma, X.; Gonzalez, F.J. CYP2E1-dependent elevation of serum cholesterol, triglycerides, and hepatic bile acids by isoniazid. Toxicol. Appl. Pharmacol. 2012, 266, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotsuka, T.; Sasaki, Y.; Hirai, S.; Yamagishi, F.; Ueno, K. Association of isoniazid-metabolizing enzyme genotypes and isoniazid-induced hepatotoxicity in tuberculosis patients. Vivo 2011, 25, 803–812. [Google Scholar]
- Swaminathan, S.; Ramachandran, G. Role of pharmacogenomics in the treatment of tuberculosis: A review. Pharm. Pers. Med. 2012, 5, 89–98. [Google Scholar] [CrossRef]
- Wang, P.; Pradhan, K.; Zhong, X.-B.; Ma, X. Isoniazid metabolism and hepatotoxicity. Acta Pharm. Sin. B 2016, 6, 384–392. [Google Scholar] [CrossRef]
- Sarich, T.C.; Adams, S.P.; Petricca, G.; Wright, J.M. Inhibition of isoniazid-induced hepatotoxicity in rabbits by pretreatment with an amidase inhibitor. J. Pharmacol. Exp. Ther. 1999, 289, 695–702. [Google Scholar] [PubMed]
- Nicoletti, P.; Devarbhavi, H.; Goel, A.; Venkatesan, R.; Eapen, C.E.; Grove, J.I.; Zafer, S.; Bjornsson, E.; Lucena, M.I.; Andrade, R.J.; et al. Genetic Risk Factors in Drug-Induced Liver Injury Due to Isoniazid-Containing Antituberculosis Drug Regimens. Clin. Pharmacol. Ther. 2020, 109, 1125–1135. [Google Scholar] [CrossRef]
- Steele, M.A.; Burk, R.F.; DesPrez, R.M. Toxic Hepatitis with Isoniazid and Rifampin: A Meta-analysis. Chest 1991, 99, 465–471. [Google Scholar] [CrossRef]
- Sarma, G.R.; Immanuel, C.; Kailasam, S.; Narayana, A.S.; Venkatesan, P. Rifampin-induced release of hydrazine from isoniazid. A possible cause of hepatitis during treatment of tuberculosis with regimens containing isoniazid and rifampin. Am. Rev. Respir. Dis. 1986, 133. [Google Scholar] [CrossRef]
- Abel, L.; Fellay, J.; Haas, D.W.; Schurr, E.; Srikrishna, G.; Urbanowski, M.; Chaturvedi, N.; Srinivasan, S.; Johnson, D.H.; Bishai, W.R. Genetics of human susceptibility to active and latent tuberculosis: Present knowledge and future perspectives. Lancet Infect. Dis. 2018, 18, e64–e75. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.A.; Gorski, J.C.; Jones, D.R.; Hall, S.D.; Flockhart, D.A.; Desta, Z. The Cytochrome P450 2B6 (CYP2B6) Is the Main Catalyst of Efavirenz Primary and Secondary Metabolism: Implication for HIV/AIDS Therapy and Utility of Efavirenz as a Substrate Marker of CYP2B6 Catalytic Activity. Experiment 2003, 306, 287–300. [Google Scholar] [CrossRef]
- Luetkemeyer, A.F.; Rosenkranz, S.L.; Lu, D.; Grinsztejn, B.; Sanchez, J.; Ssemmanda, M.; Sanne, I.; McIlleron, H.; Havlir, D.V.; Haas, D.W. Combined Effect of CYP2B6 and NAT2 Genotype on Plasma Efavirenz Exposure during Rifampin-based Antituberculosis Therapy in the STRIDE Study. Clin. Infect. Dis. 2015, 60, 1860–1863. [Google Scholar] [CrossRef]
- Kwara, A.; Lartey, M.; Sagoe, K.W.; Court, M.H. Paradoxically elevated efavirenz concentrations in HIV/tuberculosis-coinfected patients with CYP2B6 516TT genotype on rifampin-containing antituberculous therapy. Aids 2011, 25, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.E.; Denti, P.; Martinson, N.; Cohn, S.; Mashabela, F.; Hoffmann, J.; Haas, D.W.; Hull, J.; Msandiwa, R.; Castel, S.; et al. Pharmacokinetics of Efavirenz and Treatment of HIV-1 among Pregnant Women with and without Tuberculosis Coinfection. J. Infect. Dis. 2014, 211, 197–205. [Google Scholar] [CrossRef]
- DI Iulio, J.; Fayet, A.; Arab-Alameddine, M.; Rotger, M.; Lubomirov, R.; Cavassini, M.; Furrer, H.; Günthard, H.; Colombo, S.; Csajka, C.; et al. In vivo analysis of efavirenz metabolism in individuals with impaired CYP2A6 function. Pharm. Genom. 2009, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, L.; Dahl, M.-L.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, J.P.; Peter, A.P.; Shaman, J.A. Consequences of CYP2D6 Copy-Number Variation for Pharmacogenomics in Psychiatry. Front. Psychiatry 2019, 10, 432. [Google Scholar] [CrossRef]
- Zanger, U.M. Genetic variability of CYP2D6: Basic and clinical aspects. Future Med. 2014, 28–41. [Google Scholar] [CrossRef]
- Elewa, H.; Wilby, K.J. A Review of Pharmacogenetics of Antimalarials and Associated Clinical Implications. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Bains, R.K. African variation at Cytochrome P450 genes. Evol. Med. Public Health 2013, 2013, 118–134. [Google Scholar] [CrossRef]
- Adehin, A.; Igbinoba, S.I.; Soyinka, J.O.; Onyeji, C.O.; Babalola, C.P.; Bolaji, O.O. Pharmacokinetic Parameters of Quinine in Healthy Subjects and in Patients with Uncomplicated Malaria in Nigeria: Analysis of Data using a Population Approach. Curr. Ther. Res. 2019, 91, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Silvino, A.C.R.; Kano, F.S.; Costa, M.A.; Fontes, C.J.F.; Soares, I.S.; de Brito, C.F.A.; Carvalho, L.H.; Sousa, T.N. Novel Insights into Plasmodium vivax Therapeutic Failure: CYP2D6 Activity and Time of Exposure to Malaria Modulate the Risk of Recurrence. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Mukonzo, J.K.; Waako, P.; Ogwal-Okeng, J.; Gustafsson, L.L.; Aklillu, E. Genetic Variations in ABCB1 and CYP3A5 as well as Sex Influence Quinine Disposition Among Ugandans. Ther. Drug Monit. 2010, 32, 346–352. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Liu, J.-P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef]
- Mirghani, R.A.; Sayi, J.; Aklillu, E.; Allqvist, A.; Jande, M.; Wennerholm, A.; Eriksen, J.; Herben, V.M.; Jones, B.C.; Gustafsson, L.L.; et al. CYP3A5 genotype has significant effect on quinine 3-hydroxylation in Tanzanians, who have lower total CYP3A activity than a Swedish population. Pharm. Genom. 2006, 16, 637–645. [Google Scholar] [CrossRef]
- Soyinka, J.O.; Nnadi, C.O.; Onyeji, C.O. Insights and Current Perspectives on Pharmacogenomics of Antimalarial Drugs. Res. Sq. 2010. [Google Scholar] [CrossRef]
- McLeay, S.C.; Vis, P.; van Heeswijk, R.P.G.; Green, B. Population Pharmacokinetics of Bedaquiline (TMC207), a Novel Antituberculosis Drug. Antimicrob. Agents Chemother. 2014, 58, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.W.; Abdelwahab, M.T.; van Beek, S.W.; Baker, P.; Maartens, G.; Bradford, Y.; Ritchie, M.D.; Wasserman, S.; Meintjes, G.; Beeri, K.; et al. Pharmacogenetics of Between-Individual Variability in Plasma Clearance of Bedaquiline and Clofazimine in South Africa. J. Infect. Dis. 2022, 226, 147–156. [Google Scholar] [CrossRef]
- Hustert, E.; Haberl, M.; Burk, O.; Wolbold, R.; He, Y.-Q.; Klein, K.; Nuessler, A.C.; Neuhaus, P.; Klattig, J.; Eiselt, R.; et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001, 11, 773–779. [Google Scholar] [CrossRef]
- Lamba, J.; Hebert, J.M.; Schuetz, E.G.; Klein, T.E.; Altman, R.B. PharmGKB summary. Pharmacogenetics Genom. 2012, 22, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.-J.; Wu, G.; He, H.-Y.; Chen, W.; Zou, Y.-S.; Li, Q.; Zhong, L.; Huang, Y.-M.; Deng, C.-L. The association between CYP2E1 polymorphisms and hepatotoxicity due to anti-tuberculosis drugs: A meta-analysis. Infect. Genet. Evol. 2014, 24, 34–40. [Google Scholar] [CrossRef]
- Cai, Y.; Yi, J.; Zhou, C.; Shen, X. Pharmacogenetic Study of Drug-Metabolising Enzyme Polymorphisms on the Risk of Anti-Tuberculosis Drug-Induced Liver Injury: A Meta-Analysis. PLoS ONE 2012, 7, e47769. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Yang, T.; Wang, Y.; Tang, N. CYP2E1 RsaI/PstI polymorphism and risk of anti-tuberculosis drug-induced liver injury: A meta-analysis [Review article]. Int. J. Tuberc. Lung Dis. 2012, 16, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-J.; Wang, Y.; Niu, T.; Lu, W.-X.; Sandford, A.J.; He, J.-Q. Update meta-analysis of the CYP2E1 RsaI/PstI and DraI polymorphisms and risk of antituberculosis drug-induced hepatotoxicity: Evidence from 26 studies. J. Clin. Pharm. Ther. 2016, 41, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hwang, S.J.; Park, J.Y.; Chung, E.K.; I Lee, J. Association of genetic polymorphisms of CYP2E1, NAT2, GST and SLCO1B1 with the risk of anti-tuberculosis drug-induced liver injury: A systematic review and meta-analysis. BMJ Open 2019, 9, e027940. [Google Scholar] [CrossRef]
- Huang, Y.; Chern, H.; Su, W.; Wu, J.; Chang, S.; Chiang, C.; Chang, F.; Lee, S. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology 2003, 37, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-W.; Lv, X.-Z.; Zhang, Y.; Wu, S.-S.; Yang, Z.-R.; Xia, Y.-Y.; Tu, D.-H.; Deng, P.-Y.; Ma, Y.; Chen, D.-F.; et al. CYP2E1, GSTM1 and GSTT1genetic polymorphisms and susceptibility to antituberculosis drug-induced hepatotoxicity: A nested case-control study. J. Clin. Pharm. Ther. 2012, 37, 588–593. [Google Scholar] [CrossRef]
- Chamorro, J.G.; Castagnino, J.P.; Musella, R.M.; Nogueras, M.; Aranda, F.M.; Frías, A.; Visca, M.; Aidar, O.; Perés, S.; de Larrañaga, G.F. Sex, ethnicity, and slow acetylator profile are the major causes of hepatotoxicity induced by antituberculosis drugs. J. Gastroenterol. Hepatol. 2013, 28, 323–328. [Google Scholar] [CrossRef]
- Xiang, Y.; Ma, L.; Wu, W.; Liu, W.; Li, Y.; Zhu, X.; Ma, J.; Cao, M.; Wang, Q.; Yao, X.; et al. The Incidence of Liver Injury in Uyghur Patients Treated for TB in Xinjiang Uyghur Autonomous Region, China, and Its Association with Hepatic Enzyme Polymorphisms NAT2, CYP2E1, GSTM1 and GSTT1. PLoS ONE 2014, 9, e85905. [Google Scholar] [CrossRef]
- Yang, R.; Liu, H.; Chen, Z.; Qi, T.; Zhang, Z.; Qu, Q. Clinical pharmacists use CYP2C19 genotyping test to guide individual medication therapy of Clopidogrel and to evaluate the efficacy of treatment. Chin. J. Clin. Pharmacol. Ther. 2019, 24, 938. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, X.; Li, Y.; Zhu, L.; Li, S.; Zheng, G.; Ren, Q.; Xiao, Y.; Feng, F. Correlation of CpG Island Methylation of the Cytochrome P450 2E1/2D6 Genes with Liver Injury Induced by Anti-Tuberculosis Drugs: A Nested Case-Control Study. Int. J. Environ. Res. Public Health 2016, 13, 776. [Google Scholar] [CrossRef]
- Wei, Y.; Huai, C.; Zhou, C.; Gao, Y.; Chen, L.; Zhou, W.; Wei, M.; Qin, S. A methylation functional detection hepatic cell system validates correlation between DNA methylation and drug-induced liver injury. Pharm. J. 2020, 20, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Deshpande, O.; Roseman, C.C.; Rosenberg, N.A.; Feldman, M.W.; Cavalli-Sforza, L.L. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA 2005, 102, 15942–15947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandrini, M.; Asfaha, S.; Dodgen, T.M.; Warnich, L.; Pepper, M.S. Cytochrome P450 pharmacogenetics in African populations. Drug Metab. Rev. 2013, 45, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Dandara, C.; Swart, M.; Mpeta, B.; Wonkam, A.; Masimirembwa, C. Cytochrome P450 pharmacogenetics in African populations: Implications for public health. Expert Opin. Drug Metab. Toxicol. 2014, 10, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lauschke, V.M. The genetic landscape of major drug metabolizing cytochrome P450 genes—An updated analysis of population-scale sequencing data. Pharm. J. 2022, 22, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Fukami, T.; Nakajima, M.; Yoshida, R.; Tsuchiya, Y.; Fujiki, Y.; Katoh, M.; McLeod, H.L.; Yokoi, T. A novel polymorphism of human gene has an amino acid substitution (V365M) that decreases enzymatic activity in vitro and in vivo. Clin. Pharmacol. Ther. 2004, 76, 519–527. [Google Scholar] [CrossRef]
- Lewis, D.; Dickins, M.; Lake, B.; Eddershaw, P.; Tarbit, M.; Goldfarb, P. Molecular modelling of the human cytochrome P450 isoform CYP2A6 and investigations of CYP2A substrate selectivity. Toxicology 1999, 133, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.K.; Mwenifumbo, J.C.; Zhao, B.; Gillam, E.; Tyndale, R.F. A novel CYP2A6 allele, CYP2A6*23, impairs enzyme function in vitro and in vivo and decreases smoking in a population of Black-African descent. Pharm. Genom. 2008, 18, 67–75. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Nokura, R.; Hosono, H.; Hiratsuka, M.; Ishikawa, Y.; Kurimoto, E.; Oda, A. Deciphering Structural Alterations Associated with Activity Reductions of Genetic Polymorphisms in Cytochrome P450 2A6 Using Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 10119. [Google Scholar] [CrossRef]
- Zanger, U.M.; Klein, K. Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): Advances on polymorphisms, mechanisms, and clinical relevance. Front. Genet. 2013, 4, 24. [Google Scholar] [CrossRef]
- Honda, M.; Muroi, Y.; Tamaki, Y.; Saigusa, D.; Suzuki, N.; Tomioka, Y.; Matsubara, Y.; Oda, A.; Hirasawa, N.; Hiratsuka, M. Functional Characterization of CYP2B6 Allelic Variants in Demethylation of Antimalarial Artemether. Drug Metab. Dispos. 2011, 39, 1860–1865. [Google Scholar] [CrossRef]
- Klein, K.; Lang, T.; Saussele, T.; Barbosa-Sicard, E.; Schunck, W.-H.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. Genetic variability of CYP2B6 in populations of African and Asian origin: Allele frequencies, novel functional variants, and possible implications for anti-HIV therapy with efavirenz. Pharm. Genom. 2005, 15, 861–873. [Google Scholar] [CrossRef]
- Arnold, W.R.; Zelasko, S.; Meling, D.D.; Sam, K.; Das, A. Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling. Int. J. Mol. Sci. 2019, 20, 4626. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Miteva, M.A. Advances in Molecular Modeling of Human Cytochrome P450 Polymorphism. J. Mol. Biol. 2013, 425, 3978–3992. [Google Scholar] [CrossRef]
- Parikh, S.J.; Kamat, S.; Phillips, M.; Boyson, S.P.; Yarbrough, T.; Davie, D.; Zhang, Q.; Glass, K.C.; Shah, M.B. Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison. Int. J. Mol. Sci. 2021, 22, 10206. [Google Scholar] [CrossRef]
- Gyulkhandanyan, A.; Rezaie, A.R.; Roumenina, L.; Lagarde, N.; Fremeaux-Bacchi, V.; Miteva, M.; Villoutreix, B.O. Analysis of protein missense alterations by combining sequence- and structure-based methods. Mol. Genet. Genom. Med. 2020, 8, e1166. [Google Scholar] [CrossRef]
- Blaisdell, J.; Jorge-Nebert, L.F.; Coulter, S.; Ferguson, S.S.; Lee, S.-J.; Chanas, B.; Xi, T.; Mohrenweiser, H.; Ghanayem, B.; A Goldstein, J. Discovery of new potentially defective alleles of human CYP2C9. Pharmacogenetics 2004, 14, 527–537. [Google Scholar] [CrossRef]
- Blaisdell, J.; Mohrenweiser, H.; Jackson, J.; Ferguson, S.; Coulter, S.; Chanas, B.; Xi, T.; Ghanayem, B.; A Goldstein, J. Identification and functional characterization of new potentially defective alleles of human CYP2C19. Pharmacogenetics 2002, 12, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.-Q.; Wang, J.-F.; Chen, C.; Li, Y.; Chou, K.-C. Molecular Modeling of Two CYP2C19 SNPs and Its Implications for Personalized Drug Design. Protein Pept. Lett. 2008, 15, 27–32. [Google Scholar] [CrossRef]
- Arendse, L.B. Exploring the Effects of Polymorphic Variation on the Stability and Function of Human Cytochrome P450 Enzymes In Silico And In Vitro. Ph.D. Thesis, University of Cape Town, Cape Town, South Africa, 2014. [Google Scholar]
- Dong, A.N.; Ahemad, N.; Pan, Y.; Palanisamy, U.D.; Yiap, B.C.; Ong, C.E. Functional and structural characterisation of common cytochrome P450 2D6 allelic variants—Roles of Pro34 and Thr107 in catalysis and inhibition. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Nakagome, I.; Yamaotsu, N.; Gouda, H.; Hirono, S. In Silieo Study on the Inhibitory Interaction of Drugs with Wild-type CYP2D6.1 and the Natural Variant CYP2D6.17. Drug Metab. Pharmacokinet. 2014, 29, 52–60. [Google Scholar] [CrossRef]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [CrossRef]
- Wennerholm, A.; Johansson, I.; Hidestrand, M.; Bertilsson, L.; Gustafsson, L.L.; Ingelman-Sundberg, M. Characterization of the CYP2D6*29 allele commonly present in a black Tanzanian population causing reduced catalytic activity. Pharmacogenetics 2001, 11, 417–427. [Google Scholar] [CrossRef]
- Fang, P.; Tang, P.-F.; Xu, R.-A.; Zheng, X.; Wen, J.; Bao, S.-S.; Cai, J.-P.; Hu, G.-X. Functional assessment of CYP3A4 allelic variants on lidocaine metabolism in vitro. Drug Des. Dev. Ther. 2017, 11, 3503–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimalarial Drug | CYP Enzyme Metabolizer | Mechanism of Action | References |
|---|---|---|---|
| Amodiaquine | CYP2C8 | Metabolised to desethylamodiaquine (DEAQ)—likely to proceed in two steps, a hydrogen abstraction and hydroxylation at the adjacent carbon, forming an unstable carbinolamide that rapidly hydrolyzes to DEAQ and acetaldehyde. | [116] |
| Arteether | CYP2B6, CYP3A4, CYP3A5 | Arteether is deethylated to dihydroartemisinin (DHA), the main bioactive metabolite of atermisinin and its derivatives. | [120] |
| Artelinic acid | CYP3A4, CYP3A5 | Artelinic acid is O-debenzylated to DHA | [120,121] |
| Artemether | CYP3A4, CYP1A2, CYP2B6 | Artemether is demethylated to the bioactive metabolite DHA | [122,123,124] |
| Artemisinin | CYP2A6, CYP2B6, CYP3A4 | Artemisinin is not itself metabolized to DHA but acts as the primary antimalarial. Upon reaction with Fe2+ it is converted first into oxygen centered free radicals derived by reductive cleavage of its peroxide bridge, which are then converted into carbon centered free radicals by intramolecular hydrogen abstraction from CH2 groups on the periphery of the artemisinin by the O centered radicals. | [116,125,126] |
| Artesunate | CYP2A6, CYP2B6 | Rapidly hydrolyzed to the bioactive metabolite DHA | [116,125,126,127] |
| Chloroquine | CYP2C8, CYP2C19, CYP3A4, CYP2D6, CYP3A4, CYP3A5 | Chloroquine is dealkylated into N-desethylchloroquine (DCQ) and N-bis-desethylchloroquine (BDCQ), with DCQ being the major metabolite. | [113,128,129] |
| Dapsone | CYP2C9, CYP3A4 | Unlike other antimalarials, the first metabolizing enzyme of dapsone is N-acetyltransferase which hydrolyses the drug to the active form monoacetyl dapsone. CYP enzymes on the other hand hydrolyze the drug to its N-hydroxy metabolites dapsone hydroxylamine and monoacetyl hydroxylamine which are harmful hemolytic metabolites. | [116,117,119,130,131] |
| Halofantrine and Lumefantrine | CYP3A4, CYP3A5 | Halofantrine undergoes desbutylation to N-desbutyl-halofantrine which possesses some antimalarial activity while lumefantrine is metabolized to desbutyl-lumefantrine and excreted via bile and faces. | [115,132] |
| Mefloquine | CYP1A2, CYP3A4 | Metabolized into carboxymefloquine metabolite which has little or no antimalarial activity. | [121,133,134,135] |
| Primaquine | CYP1A2, CYP3A4, CYP2D6 | Three possible pathways exist for primaquine metabolism and that involving CYP enzymes is hydroxylation at different positions on the quinoline ring, with mono-, di-, or even tri-hydroxylations possible, and subsequent glucuronide conjugation of the hydroxylated metabolites. The main metabolite carboxyprimaquine comes about through a monoamine oxidase catalyzed oxidative deamination. | [116,136] |
| Proguanil | CYP2C19 | Oxidative metabolized to cycloguanil, which is the active form of the drug | [116,121,137] |
| Quinine | CYP3A4 | Undergoes hydroxylation to the main metabolite 3-hydroxiquinine | [28,121,138,139] |
| CYP Enzyme | Antimalarial/Antituberculosis Drug Metabolized | Allele | Frequency in African Populations (%) | Amino acid Mutation Position | Functional Consequence | Residue/Mutation Notes |
|---|---|---|---|---|---|---|
| CYP2A6 | Artemisinin, artesunate | CYP2A6*17 | 10.9 | V365M | - | Located in CYP2A6 substrate recognition site SRS-5 forming part of the active site region and some side chains that point into the heme pocket, suggesting this residue may be important for substrate specificity. In vitro enzyme assays and metabolism studies showed no effect of the mutation on the stability of the enzyme. Both valine and methionine are non-polar hydrophobic residues; however, methionine is larger and contains a sulfur atom [231]. |
| CYP2A6*23 | 1.4 | R203C | Decreased | Residue located on α-helix F in substrate recognition site 2/3. Change from large basic residue to medium sized polar residue. Molecular modeling suggests Arg203 could orient important binding residue Phe209 [232,233]. | ||
| CYP2A6*25 | 1.4 | F118L | - | During molecular dynamics simulation, the F118L mutant side chain moves away from the heme and affects secondary structure formation and interaction with heme and substrates [234]. | ||
| CYP2A6*28 | 1.5 | N418D E419D | - | N418D and E419D cause a structural change in the substrate access channel and the substrate binding site [234]. | ||
| CYP2B6 | Artemisinin, artesunate, arteether | CYP2B6*6 | 32 | Q172H; K262R | Decreased | Q172H and K262R are not located at the active site and have not been identified in substrate recognition sites [235]. |
| CYP2B6*18 | 7 | I328T | Inactive | Results in no detectable protein or activity in vitro [236]. Designated as a null allele [237]. The only commonly found inactive CYP2B6 missense mutation, and highly restricted to Africa [230]. | ||
| CYP2C8 | Chloroquine, amodiaquine, bedaquiline | CYP2C8*2 | 15.2 | I269F | Decreased | Residue located on enzyme surface. Larger residue change. Possible effect on enzyme folding and interaction with cytochrome P450 reductase [238]. |
| CYP 2C9 | Dapsone | CYP 2C9*5 | 1.1 | D360E | Decreased | In molecular dynamics simulations, D360E broke the hydrogen bond between D360 and S478 (helix K and loop β4) leading to local structure destabilization. Glutamic acid is larger than aspartic acid [239]. |
| CYP 2C9*8 | 6 | R150H | Decreased | R150 is highly conserved. Located on protein surface at D-helix region, away from the active site. The structure of the CYP2C9*8-losartan complex has ~60° rotation of the H150 sidechain in an alternate conformation compared to the sidechain of R150 [240]. Possible involvement of R150H in the salt bridge network with the neighboring residues, suggesting the change to histidine at this solvent-exposed region may no longer coordinate similar ionic and electrostatic interactions and may result in destabilization of the structure [241]. Possible that this change could influence reductase binding [242]. | ||
| CYP 2C9*9 | 7.5 | H251R | Normal | H251R is located near the C-terminal end of helix G and hydrogen bonds to D262 on helix H. Arginine has a longer side chain than histidine [242]. | ||
| CYP 2C19 | Proguanil, quinine, dapsone, bedaquiline | CYP 2C19*9 | 1.3 | R144H I331V | Decreased | The R144H mutation could affect enzyme structure and function. The conserved arginine is located in the D helix and is part of a complex salt bridge with conserved Ser180 in the E helix and the backbone of the turn before helix H. The arginine could help stabilize the structure of the enzyme and may also be a part of the hinge for the F–G loop [243]. Docking experiments suggest that I331 is involved in ligand binding. The I331V mutant has differing lipophilicity in the binding pocket or active cavity [239,244]. |
| CYP 2C19*13 | 1.8 | I331V R410C | Normal | Residue 410 located on enzyme surface. Change from large basic residue to medium sized polar residue. Mutation has neutral effect on enzyme activity. | ||
| CYP 2C19*15 | 1.9 | I19L I331V | Normal | The I19L mutation with residue 19 is part of signal-anchor sequence, located in truncated N-terminal region. Mutation has neutral effect [245]. | ||
| CYP 2D6 | Chloroquine | CYP 2D6*2 | 22.5 | R296C S486T | Normal | R296C is located in substrate recognition site 4 (I-helix) important for catalytic proton delivery. Kinetics data indicate that R296C in CYP2D6*2 causes enhanced ligand binding possibly due to morphological changes Change from medium-sized basic residue to small polar residue. [246]. S486T mutation is located in substrate recognition site (β4-2 sheet) and is very close to active site and heme [239]. Kinetics data indicate that S486T in CYP2D6*2 causes enhanced ligand binding possibly due to morphological changes [246]. |
| CYP 2D6*17 | 20.5 | T107I R296C S486T | Decreased | T107I is located in substrate recognition site 1 (B′-helix). Substitution of hydrophilic Threonine to hydrophobic Isoleucine. Threonine has alcoholic side chain pointing down towards heme, whereas Isoleucine long aliphatic side chain points away from active site [246]. Causes changes in hydrogen bonds with surrounding residues within the active site cavity [247]. | ||
| CYP 2D6*29 | 8.9 | V136I R296C V338M S486T | Decreased | V136 is located between substrate recognition sites 1 and 2 in helix C [248]. In contact with cytochrome P450 reductase. Neutral effect on protein stability [245]. V338M is located between substrate recognition sites 4 and 5 at the end of helix J [248]. Neutral effect on protein stability [245]. Expression studies reveal that both the V136I and the V338M mutations affect catalytic activity, and the effect is stronger when present together as seen in CYP2D6*29. Both valine and methionine are non-polar hydrophobic residues; however, methionine is larger and contains a sulfur atom [249]. | ||
| CYP3A4 | Quinine, quinidine, chloroquine, mefloquine, primaquine, halofantrine, lumefantrine, dapsone, artemisinin, artemether, arteether, artelinic, acid, delaminid, bedaquiline | CYP3A4*15 | 2.6 | R162Q | - | Mutation related to rapid metabolism of quinine in vitro. Change from large basic residue to medium-sized polar hydrophilic residue [250]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamboko, C.R.; Veldman, W.; Tata, R.B.; Schoeberl, B.; Tastan Bishop, Ö. Human Cytochrome P450 1, 2, 3 Families as Pharmacogenes with Emphases on Their Antimalarial and Antituberculosis Drugs and Prevalent African Alleles. Int. J. Mol. Sci. 2023, 24, 3383. https://doi.org/10.3390/ijms24043383
Chamboko CR, Veldman W, Tata RB, Schoeberl B, Tastan Bishop Ö. Human Cytochrome P450 1, 2, 3 Families as Pharmacogenes with Emphases on Their Antimalarial and Antituberculosis Drugs and Prevalent African Alleles. International Journal of Molecular Sciences. 2023; 24(4):3383. https://doi.org/10.3390/ijms24043383
Chicago/Turabian StyleChamboko, Chiratidzo R., Wayde Veldman, Rolland Bantar Tata, Birgit Schoeberl, and Özlem Tastan Bishop. 2023. "Human Cytochrome P450 1, 2, 3 Families as Pharmacogenes with Emphases on Their Antimalarial and Antituberculosis Drugs and Prevalent African Alleles" International Journal of Molecular Sciences 24, no. 4: 3383. https://doi.org/10.3390/ijms24043383