
Secondary Degeneration Impairs Myelin Ultrastructural Development in Adulthood following Adolescent Neurotrauma in the Rat Optic Nerve
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
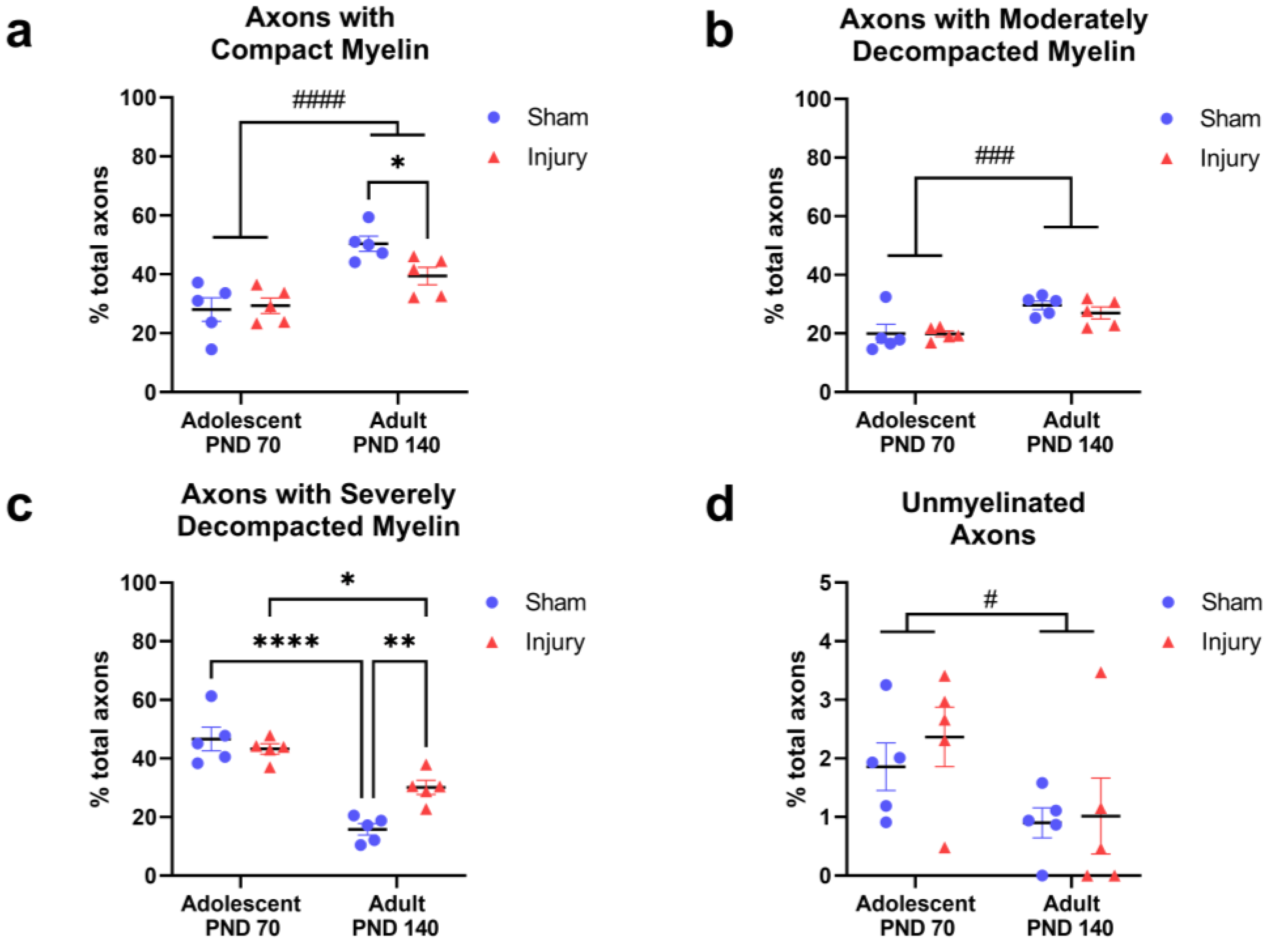
2.1. Injury in Adolescence Was Associated with a Lower Percentage of Axons with Compact Myelin and a Higher Percentage of Axons with Severely Decompacted Myelin at Adulthood
2.2. Injury in Adolescence Prevented Development of Thicker, more Compact Myelin in Adulthood
2.3. The Diameter of Axons with Moderate Myelin Decompaction Increased with Age, but Not Injury
2.4. The Relationship between Axon Diameter and Myelin Thickness Deteriorated in Adulthood after Injury in Early Adolescence
3. Discussion
3.1. Injury in Adolescence Impaired Myelin Maturation
3.2. Ultrastructure of Myelin Matured between Adolescence and Adulthood
3.3. Overt Myelin Ultrastructural Changes Were Not Observed 2 Weeks after Injury
4. Materials and Methods
4.1. Animals
4.2. Optic Nerve Partial Transection Surgery
4.3. Tissue Preparation
4.4. Image Acquisition
4.5. Ultrastructural Analysis of Myelinated Axons
4.6. Statistical Analyses and Data Presentation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Giedd, J.N.; Blumenthal, J.; Jeffries, N.O.; Castellanos, F.X.; Liu, H.; Zijdenbos, A.; Paus, T.; Evans, A.C.; Rapoport, J.L. Brain development during childhood and adolescence: A longitudinal MRI study. Nat. Neurosci. 1999, 2, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Beaulieu, C. Longitudinal Development of Human Brain Wiring Continues from Childhood into Adulthood. J. Neurosci. 2011, 31, 10937–10947. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Gee, M.; Camicioli, R.; Wieler, M.; Martin, W.; Beaulieu, C. Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage 2012, 60, 340–352. [Google Scholar] [CrossRef]
- Corrigan, N.M.; Yarnykh, V.L.; Hippe, D.S.; Owen, J.P.; Huber, E.; Zhao, T.C.; Kuhl, P.K. Myelin development in cerebral gray and white matter during adolescence and late childhood. NeuroImage 2021, 227, 117678. [Google Scholar] [CrossRef] [PubMed]
- Barnea-Goraly, N.; Menon, V.; Eckert, M.; Tamm, L.; Bammer, R.; Karchemskiy, A.; Dant, C.C.; Reiss, A.L. White matter development during childhood and adolescence: A cross-sectional diffusion tensor imaging study. Cereb. Cortex 2005, 15, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Ashtari, M.; Cervellione, K.L.; Hasan, K.M.; Wu, J.; McIlree, C.; Kester, H.; Ardekani, B.A.; Roofeh, D.; Szeszko, P.R.; Kumra, S. White matter development during late adolescence in healthy males: A cross-sectional diffusion tensor imaging study. NeuroImage 2007, 35, 501–510. [Google Scholar] [CrossRef]
- Giorgio, A.; Watkins, K.E.; Chadwick, M.; James, S.; Winmill, L.; Douaud, G.; De Stefano, N.; Matthews, P.M.; Smith, S.M.; Johansen-Berg, H.; et al. Longitudinal changes in grey and white matter during adolescence. NeuroImage 2010, 49, 94–103. [Google Scholar] [CrossRef]
- Beaulieu, C. The basis of anisotropic water diffusion in the nervous system—A technical review. NMR Biomed. 2002, 15, 435–455. [Google Scholar] [CrossRef]
- Sowell, E.R.; Thompson, P.M.; Holmes, C.J.; Jernigan, T.L.; Toga, A.W. In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nat. Neurosci. 1999, 2, 859–861. [Google Scholar] [CrossRef]
- Vanes, L.D.; Moutoussis, M.; Ziegler, G.; Goodyer, I.M.; Fonagy, P.; Jones, P.B.; Bullmore, E.T.; Consortium, N.; Dolan, R.J. White matter tract myelin maturation and its association with general psychopathology in adolescence and early adulthood. Hum. Brain Mapp. 2020, 41, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.; Keshavan, M.; Giedd, J.N. Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci. 2008, 9, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.; de la Serna, E.; Baeza, I.; Valli, I.; Pariente, J.C.; Picado, M.; Bargalló, N.; Sugranyes, G.; Castro-Fornieles, J. Altered White Matter Integrity at Illness Onset in Adolescents With a First Episode of Psychosis. Front. Psychiatry 2022, 13, 876793. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; Song, A.W.; Sim, K. White matter abnormalities in bipolar disorder: Insights from diffusion tensor imaging studies. J. Neural. Transm. 2010, 117, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Whalen, P.J. The Structural Integrity of an Amygdala–Prefrontal Pathway Predicts Trait Anxiety. J. Neurosci. 2009, 29, 11614–11618. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, G.; Moutoussis, M.; Hauser, T.U.; Fearon, P.; Bullmore, E.T.; Goodyer, I.M.; Fonagy, P.; Jones, P.B.; Lindenberger, U.; Dolan, R.J. Childhood socio-economic disadvantage predicts reduced myelin growth across adolescence and young adulthood. Hum. Brain Mapp. 2020, 41, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.R.; McCormack, K.M.; Grand, A.P.; Sawyer, N.T.; Zhang, X.; Maestripieri, D.; Hu, X.; Sanchez, M.M. Brain white matter microstructure alterations in adolescent rhesus monkeys exposed to early life stress: Associations with high cortisol during infancy. Biol. Mood Anxiety Disord. 2013, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Tasker, R.C. Changes in White Matter Late after Severe Traumatic Brain Injury in Childhood. Dev. Neurosci. 2006, 28, 302–308. [Google Scholar] [CrossRef]
- Badhiwala, J.H.; Wilson, J.R.; Fehlings, M.G. Global burden of traumatic brain and spinal cord injury. Lancet Neurol. 2019, 18, 24–25. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Bartlett, C.A.; Evill, L.; Rodger, J.; Harvey, A.R.; Dunlop, S.A. Secondary degeneration of the optic nerve following partial transection: The benefits of lomerizine. Exp. Neurol. 2009, 216, 219–230. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Payne, S.C.; Bartlett, C.A.; Evill, L.; Harvey, A.R.; Dunlop, S.A. Secondary retinal ganglion cell death and the neuroprotective effects of the calcium channel blocker lomerizine. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5456–5462. [Google Scholar] [CrossRef] [PubMed]
- Levkovitch-Verbin, H.; Quigley, H.A.; Martin, K.R.; Zack, D.J.; Pease, M.E.; Valenta, D.F. A model to study differences between primary and secondary degeneration of retinal ganglion cells in rats by partial optic nerve transection. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3388–3393. [Google Scholar] [CrossRef]
- Staal, J.A.; Dickson, T.C.; Gasperini, R.; Liu, Y.; Foa, L.; Vickers, J.C. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 2010, 112, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75 (Suppl. 4), S24–S33. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Giacci, M.; Fitzgerald, M. Oligodendroglia Are Particularly Vulnerable to Oxidative Damage After Neurotrauma In Vivo. J. Exp. Neurosci. 2018, 12, 1179069518810004. [Google Scholar] [CrossRef]
- Matute, C.; Torre, I.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodríguez-Antigüedad, A.; Sánchez-Gómez, M.; et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef]
- Borges, K.; Ohlemeyer, C.; Trotter, J.; Kettenmann, H. AMPA/kainate receptor activation in murine oligodendrocyte precursor cells leads to activation of a cation conductance, calcium influx and blockade of delayed rectifying K+ channels. Neuroscience 1994, 63, 135–149. [Google Scholar] [CrossRef]
- Yamaya, Y.; Yoshioka, A.; Saiki, S.; Yuki, N.; Hirose, G.; Pleasure, D. Type-2 astrocyte-like cells are more resistant than oligodendrocyte-like cells against non-N-methyl-D-aspartate glutamate receptor-mediated excitotoxicity. J. Neurosci. Res. 2002, 70, 588–598. [Google Scholar] [CrossRef]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef]
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808. [Google Scholar] [CrossRef]
- Giacci, M.K.; Bartlett, C.A.; Smith, N.M.; Iyer, K.S.; Toomey, L.M.; Jiang, H.; Guagliardo, P.; Kilburn, M.R.; Fitzgerald, M. Oligodendroglia Are Particularly Vulnerable to Oxidative Damage after Neurotrauma In Vivo. J. Neurosci. 2018, 38, 6491–6504. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.C.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Chronic swelling and abnormal myelination during secondary degeneration after partial injury to a central nervous system tract. J. Neurotrauma 2011, 28, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.C.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Myelin sheath decompaction, axon swelling, and functional loss during chronic secondary degeneration in rat optic nerve. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6093–6101. [Google Scholar] [CrossRef] [PubMed]
- Savigni, D.L.; O’Hare Doig, R.L.; Szymanski, C.R.; Bartlett, C.A.; Lozić, I.; Smith, N.M.; Fitzgerald, M. Three Ca2+ channel inhibitors in combination limit chronic secondary degeneration following neurotrauma. Neuropharmacology 2013, 75, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.C.; Bartlett, C.A.; Savigni, D.L.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Early proliferation does not prevent the loss of oligodendrocyte progenitor cells during the chronic phase of secondary degeneration in a CNS white matter tract. PLoS ONE 2013, 8, e65710. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, C.R.; Chiha, W.; Morellini, N.; Cummins, N.; Bartlett, C.A.; O’Hare Doig, R.L.; Savigni, D.L.; Payne, S.C.; Harvey, A.R.; Dunlop, S.A.; et al. Paranode Abnormalities and Oxidative Stress in Optic Nerve Vulnerable to Secondary Degeneration: Modulation by 670 nm Light Treatment. PLoS ONE 2013, 8, e66448. [Google Scholar] [CrossRef]
- Chiha, W.; Bartlett, C.A.; Petratos, S.; Fitzgerald, M.; Harvey, A.R. Intravitreal application of AAV-BDNF or mutant AAV-CRMP2 protects retinal ganglion cells and stabilizes axons and myelin after partial optic nerve injury. Exp. Neurol. 2020, 326, 113167. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A. Early events of secondary degeneration after partial optic nerve transection: An immunohistochemical study. J. Neurotrauma 2010, 27, 439–452. [Google Scholar] [CrossRef]
- Donovan, V.; Kim, C.; Anugerah, A.K.; Coats, J.S.; Oyoyo, U.; Pardo, A.C.; Obenaus, A. Repeated mild traumatic brain injury results in long-term white-matter disruption. J. Cereb. Blood Flow Metab. 2014, 34, 715–723. [Google Scholar] [CrossRef]
- Chen, H.S.-M.; Holmes, N.; Liu, J.; Tetzlaff, W.; Kozlowski, P. Validating myelin water imaging with transmission electron microscopy in a rat spinal cord injury model. NeuroImage 2017, 153, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Nashmi, R.; Fehlings, M.G. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience 2001, 104, 235–251. [Google Scholar] [CrossRef]
- Anthes, D.L.; Theriault, E.; Tator, C.H. Characterization of axonal ultrastructural pathology following experimental spinal cord compression injury. Brain Res. 1995, 702, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Walker, L.; Leemans, A.; Phillips, L.; Beaulieu, C. Microstructural maturation of the human brain from childhood to adulthood. NeuroImage 2008, 40, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Seidl, A.H. Regulation of conduction time along axons. Neuroscience 2014, 276, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, R.; Boison, D.; Heinemann, U.; Stoffel, W. Decompaction of CNS myelin leads to a reduction of the conduction velocity of action potentials in optic nerve. Neurosci. Lett. 1995, 195, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Boiko, T.; Rasband, M.N.; Levinson, S.R.; Caldwell, J.H.; Mandel, G.; Trimmer, J.S.; Matthews, G. Compact Myelin Dictates the Differential Targeting of Two Sodium Channel Isoforms in the Same Axon. Neuron 2001, 30, 91–104. [Google Scholar] [CrossRef]
- Balraj, A.; Clarkson-Paredes, C.; Pajoohesh-Ganji, A.; Kay, M.W.; Mendelowitz, D.; Miller, R.H. Refinement of axonal conduction and myelination in the mouse optic nerve indicate an extended period of postnatal developmental plasticity. Dev. Neurobiol. 2022, 82, 308–325. [Google Scholar] [CrossRef]
- Ferrer, E.; Whitaker, K.J.; Steele, J.S.; Green, C.T.; Wendelken, C.; Bunge, S.A. White matter maturation supports the development of reasoning ability through its influence on processing speed. Dev. Sci. 2013, 16, 941–951. [Google Scholar] [CrossRef]
- Turken, U.; Whitfield-Gabrieli, S.; Bammer, R.; Baldo, J.V.; Dronkers, N.F.; Gabrieli, J.D.E. Cognitive processing speed and the structure of white matter pathways: Convergent evidence from normal variation and lesion studies. NeuroImage 2008, 42, 1032–1044. [Google Scholar] [CrossRef]
- Sánchez, I.; Hassinger, L.; Paskevich, P.A.; Shine, H.D.; Nixon, R.A. Oligodendroglia regulate the regional expansion of axon caliber and local accumulation of neurofilaments during development independently of myelin formation. J. Neurosci. 1996, 16, 5095–5105. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, B.L.; Lebel, R.M.; Mah, A.C.; Deoni, S.C.; Alsop, D.C.; Varma, G.; Lebel, C. A comparison of inhomogeneous magnetization transfer, myelin volume fraction, and diffusion tensor imaging measures in healthy children. NeuroImage 2018, 182, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hüppi, P.S.; Dubois, J. Diffusion tensor imaging of brain development. Semin. Fetal Neonatal Med. 2006, 11, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Oishi, K.; Faria, A.V.; Mori, S. Diffusion tensor imaging of normal brain development. Pediatr. Radiol. 2013, 43, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Kilburn, M.R.; Shaw, J.A.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Early in vivo changes in calcium ions, oxidative stress markers, and ion channel immunoreactivity following partial injury to the optic nerve. J. Neurosci. Res. 2012, 90, 606–618. [Google Scholar] [CrossRef]
- Toomey, L.M.; Bartlett, C.A.; Majimbi, M.; Gopalasingam, G.; Rodger, J.; Fitzgerald, M. Comparison of ion channel inhibitor combinations for limiting secondary degeneration following partial optic nerve transection. Exp. Brain Res. 2019, 237, 161–171. [Google Scholar] [CrossRef]
- Toomey, L.M.; Bartlett, C.A.; Gavriel, N.; McGonigle, T.; Majimbi, M.; Gopalasingam, G.; Rodger, J.; Fitzgerald, M. Comparing modes of delivery of a combination of ion channel inhibitors for limiting secondary degeneration following partial optic nerve transection. Sci. Rep. 2019, 9, 15297. [Google Scholar] [CrossRef]
- Cummins, N.; Bartlett, C.A.; Archer, M.; Bartlett, E.; Hemmi, J.M.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Changes to mitochondrial ultrastructure in optic nerve vulnerable to secondary degeneration in vivo are limited by irradiation at 670 nm. BMC Neurosci. 2013, 14, 98. [Google Scholar] [CrossRef]
- Chiang, A.C.A.; Seua, A.V.; Singhmar, P.; Arroyo, L.D.; Mahalingam, R.; Hu, J.; Kavelaars, A.; Heijnen, C.J. Bexarotene normalizes chemotherapy-induced myelin decompaction and reverses cognitive and sensorimotor deficits in mice. Acta Neuropathol. Commun. 2020, 8, 193. [Google Scholar] [CrossRef]
- Chu, P.H.; Li, H.-Y.; Chin, M.-P.; So, K.-f.; Chan, H.H. Effect of lycium barbarum (wolfberry) polysaccharides on preserving retinal function after partial optic nerve transection. PLoS ONE 2013, 8, e81339. [Google Scholar] [CrossRef]
- Levkovitch-Verbin, H.; Spierer, O.; Vander, S.; Dardik, R. Similarities and differences between primary and secondary degeneration of the optic nerve and the effect of minocycline. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 849–857. [Google Scholar] [CrossRef]
- Ghasemi, A.; Jeddi, S.; Kashfi, K. The laboratory rat: Age and body weight matter. EXCLI J. 2021, 20, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Reeves, T.M.; Smith, T.L.; Williamson, J.C.; Phillips, L.L. Unmyelinated axons show selective rostrocaudal pathology in the corpus callosum after traumatic brain injury. J. Neuropathol. Exp. Neurol. 2012, 71, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Giacci, M.K.; Bartlett, C.A.; Huynh, M.; Kilburn, M.R.; Dunlop, S.A.; Fitzgerald, M. Three dimensional electron microscopy reveals changing axonal and myelin morphology along normal and partially injured optic nerves. Sci. Rep. 2018, 8, 3979. [Google Scholar] [CrossRef]
- Kelley, B.J.; Farkas, O.; Lifshitz, J.; Povlishock, J.T. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp. Neurol. 2006, 198, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Saggu, S.K.; Chotaliya, H.P.; Blumbergs, P.C.; Casson, R.J. Wallerian-like axonal degeneration in the optic nerve after excitotoxic retinal insult: An ultrastructural study. BMC Neurosci. 2010, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Fehily, B.; Bartlett, C.A.; Lydiard, S.; Archer, M.; Milbourn, H.; Majimbi, M.; Hemmi, J.M.; Dunlop, S.A.; Yates, N.J.; Fitzgerald, M. Differential responses to increasing numbers of mild traumatic brain injury in a rodent closed-head injury model. J. Neurochem. 2019, 149, 660–678. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Carroll, R.J.; Harden, K.K.; Wu, G. Comparisons of treatment means when factors do not interact in two-factorial studies. Amino Acids 2012, 42, 2031–2035. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adult (PND 140) Relative to Adolescent (PND 70) Overall | Injury-Adolescent (PND 70) Relative to Sham-Adolescent (PND 70) | Sham-Adult (PND 140) Relative to Sham-Adolescent (PND 70) | Injury-Adult (PND 140) Relative to Sham-Adult (PND 140) | Injury-Adult (PND 140) Relative to Injury-Adolescent (PND 70) | |
| Myelin Classifications | |||||
| Axons with Compact Myelin (%) | ↑↑ | ns | ― | ↓ | ― |
| Axons with moderately decompacted myelin (%) | ↑↑ | ― | ― | ― | ― |
| Axons with severely decompacted myelin (%) | ↓↓ | ns | ↓↓ | ↑ | ↓ |
| Unmyelinated axons (%) | ↓ | ― | ― | ― | ― |
| Morphology of Axons with Compact Myelin | |||||
| Myelin Thickness (µm) | ↑↑ | ― | ↑↑ | ― | ns |
| Axon Diameter (µm) | ns | ― | ― | ― | ― |
| Fibre Diameter (µm) | ns | ― | ― | ― | ― |
| G Ratio | ns | ― | ― | ― | ― |
| Diameter of Axons with Decompacted Myelin | |||||
| Axons with Moderately Decompacted Myelin (µm) | ↑ | ― | ― | ― | ― |
| Axons with Severely Decompacted Myelin (µm) | ns | ― | ― | ― | ― |
| Axons with Completely Decompacted Myelin (µm) | ns | ― | ― | ― | ― |
| Unmyelinated Axons (µm) | ns | ― | ― | ― | ― |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lins, B.R.; Anyaegbu, C.C.; McGonigle, T.; Hellewell, S.C.; Patel, P.; Reagan, H.; Rooke-Wiesner, C.; Warnock, A.; Archer, M.; Hemmi, J.M.; et al. Secondary Degeneration Impairs Myelin Ultrastructural Development in Adulthood following Adolescent Neurotrauma in the Rat Optic Nerve. Int. J. Mol. Sci. 2023, 24, 3343. https://doi.org/10.3390/ijms24043343
Lins BR, Anyaegbu CC, McGonigle T, Hellewell SC, Patel P, Reagan H, Rooke-Wiesner C, Warnock A, Archer M, Hemmi JM, et al. Secondary Degeneration Impairs Myelin Ultrastructural Development in Adulthood following Adolescent Neurotrauma in the Rat Optic Nerve. International Journal of Molecular Sciences. 2023; 24(4):3343. https://doi.org/10.3390/ijms24043343
Chicago/Turabian StyleLins, Brittney R., Chidozie C. Anyaegbu, Terence McGonigle, Sarah C. Hellewell, Parth Patel, Harry Reagan, Cara Rooke-Wiesner, Andrew Warnock, Michael Archer, Jan M. Hemmi, and et al. 2023. "Secondary Degeneration Impairs Myelin Ultrastructural Development in Adulthood following Adolescent Neurotrauma in the Rat Optic Nerve" International Journal of Molecular Sciences 24, no. 4: 3343. https://doi.org/10.3390/ijms24043343