
Targeting and Monitoring Acute Myeloid Leukaemia with Nucleophosmin-1 (NPM1) Mutation

Abstract
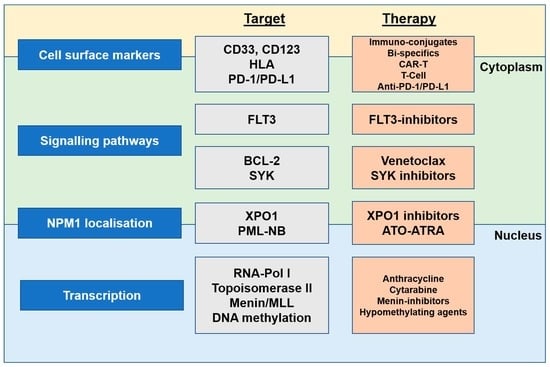
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Functional Role of NPM1
2.1. Genetic Characteristics
2.2. Functions of NPM1
2.2.1. Ribosome Biogenesis
2.2.2. RNA Processing
2.2.3. Chromatin Remodelling
2.2.4. Regulation of Apoptosis and Cellular Growth
2.2.5. Genome Instability
3. NPM1 Mutations in AML and Other Malignancies
4. Measurable-residual monitoring in NPM1-mutated AML
4.1. Conventional Technologies for MRD
4.2. Advanced Technologies for NPM1 MRD Monitoring
5. Targeting NPM1-Mutated AML
5.1. Current Agents or Standard Treatment
5.1.1. Anthracycline
5.1.2. Cytarabine
5.1.3. FLT3 Inhibitors
5.1.4. Gemtuzumab Ozogamicin
5.1.5. Venetoclax
5.1.6. Hypomethylating Drugs
5.1.7. Actinomycin D
5.2. Novel Agents and Future Directions
5.2.1. Arsenic Trioxide (ATO) and All-Trans Retinoic Acid (ATRA)
5.2.2. XPO1 Inhibitors
5.2.3. Menin Inhibitors
5.2.4. HLA-Dependent T-Cell Immunotherapy
5.2.5. CAR-T Therapy
5.2.6. Immune Checkpoint Inhibition
5.2.7. SYK Inhibitor
5.2.8. Stress Inducing AML
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Umekawa, H.; Chang, J.H.; Correia, J.J.; Wang, D.; Wingfield, P.T.; Olson, M.O. Nucleolar protein B23: Bacterial expression, purification, oligomerization and secondary structures of two isoforms. Cell. Mol. Biol. Res. 1993, 39, 635–645. [Google Scholar]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef]
- Martelli, M.P.; Rossi, R.; Venanzi, A.; Meggendorfer, M.; Perriello, V.M.; Martino, G.; Spinelli, O.; Ciurnelli, R.; Varasano, E.; Brunetti, L.; et al. Novel NPM1 exon 5 mutations and gene fusions leading to aberrant cytoplasmic nucleophosmin in AML. Blood 2021, 138, 2696–2701. [Google Scholar] [CrossRef]
- Duployez, N.; Chebrek, L.; Helevaut, N.; Fournier, E.; Bemba, M.; Caillault, A.; Geffroy, S.; Preudhomme, C. A novel type of NPM1 mutation characterized by multiple internal tandem repeats in a case of cytogenetically normal acute myeloid leukemia. Haematologica 2018, 103, e575–e577. [Google Scholar] [CrossRef]
- Kelemen, K. The Role of Nucleophosmin 1 (NPM1) Mutation in the Diagnosis and Management of Myeloid Neoplasms. Life 2022, 12, 109. [Google Scholar] [CrossRef]
- Hindley, A.; Catherwood, M.A.; McMullin, M.F.; Mills, K.I. Significance of NPM1 Gene Mutations in AML. Int. J. Mol. Sci. 2021, 22, 10400. [Google Scholar] [CrossRef]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef]
- Hingorani, K.; Szebeni, A.; Olson, M.O. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 2000, 275, 24451–24457. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Grace, C.R.; Buljan, M.; Yun, M.K.; Pytel, N.J.; Satumba, J.; Nourse, A.; Park, C.G.; Madan Babu, M.; White, S.W.; et al. Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc. Natl. Acad. Sci. USA 2014, 111, 4466–4471. [Google Scholar] [CrossRef]
- McBride, K.M.; McDonald, C.; Reich, N.C. Nuclear export signal located within theDNA-binding domain of the STAT1transcription factor. Embo. J. 2000, 19, 6196–6206. [Google Scholar] [CrossRef]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef]
- Wang, A.J.; Han, Y.; Jia, N.; Chen, P.; Minden, M.D. NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia 2020, 34, 1278–1290. [Google Scholar] [CrossRef]
- Olausson, K.H.; Nistér, M.; Lindström, M.S. Loss of nucleolar histone chaperone NPM1 triggers rearrangement of heterochromatin and synergizes with a deficiency in DNA methyltransferase DNMT3A to drive ribosomal DNA transcription. J. Biol. Chem. 2014, 289, 34601–34619. [Google Scholar] [CrossRef]
- Cela, I.; Di Matteo, A.; Federici, L. Nucleophosmin in its interaction with ligands. Int. J. Mol. Sci. 2020, 21, 4885. [Google Scholar] [CrossRef]
- Chan, W.Y.; Liu, Q.R.; Borjigin, J.; Busch, H.; Rennert, O.M.; Tease, L.A.; Chan, P.K. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28, 1033–1039. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Stanley, C.B.; Nourse, A.; Onuchic, P.L.; Banerjee, P.R.; Phillips, A.H.; Park, C.G.; Deniz, A.A.; Kriwacki, R.W. Self-interaction of NPM1 modulates multiple mechanisms of liquid-liquid phase separation. Nat. Commun. 2018, 9, 842. [Google Scholar] [CrossRef]
- Smetana, K. Structural features of nucleoli in blood, leukemic, lymphoma and myeloma cells. Eur. J. Histochem. 2002, 46, 125–132. [Google Scholar] [CrossRef]
- Hisaoka, M.; Nagata, K.; Okuwaki, M. Intrinsically disordered regions of nucleophosmin/B23 regulate its RNA binding activity through their inter- and intra-molecular association. Nucleic Acids Res. 2014, 42, 1180–1195. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [CrossRef]
- Okuwaki, M.; Saito, S.; Hirawake-Mogi, H.; Nagata, K. The interaction between nucleophosmin/NPM1 and the large ribosomal subunit precursors contribute to maintaining the nucleolar structure. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118879. [Google Scholar] [CrossRef]
- Twayana, S.; Bacolla, A.; Barreto-Galvez, A.; De-Paula, R.B.; Drosopoulos, W.C.; Kosiyatrakul, S.T.; Bouhassira, E.E.; Tainer, J.A.; Madireddy, A.; Schildkraut, C.L. Translesion polymerase eta both facilitates DNA replication and promotes increased human genetic variation at common fragile sites. Proc. Natl. Acad. Sci. USA 2021, 118, e2106477118. [Google Scholar] [CrossRef]
- Ziv, O.; Zeisel, A.; Mirlas-Neisberg, N.; Swain, U.; Nevo, R.; Ben-Chetrit, N.; Martelli, M.P.; Rossi, R.; Schiesser, S.; Canman, C.E.; et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat. Commun. 2014, 5, 5437. [Google Scholar] [CrossRef]
- Chen, L.; Hu, N.; Wang, C.; Zhao, H. HOTAIRM1 knockdown enhances cytarabine-induced cytotoxicity by suppression of glycolysis through the Wnt/β-catenin/PFKP pathway in acute myeloid leukemia cells. Arch. Biochem. Biophys. 2020, 680, 108244. [Google Scholar] [CrossRef]
- Gourvest, M.; De Clara, E.; Wu, H.-C.; Touriol, C.; Meggetto, F.; De Thé, H.; Pyronnet, S.; Brousset, P.; Bousquet, M. A novel leukemic route of mutant NPM1 through nuclear import of the overexpressed long noncoding RNA LONA. Leukemia 2021, 35, 2784–2798. [Google Scholar] [CrossRef]
- Karsenti, E.; Newport, J.; Kirschner, M. Respective roles of centrosomes and chromatin in the conversion of microtubule arrays from interphase to metaphase. J. Cell. Biol. 1984, 99, 47s–54s. [Google Scholar] [CrossRef]
- Keryer, G.; Di Fiore, B.; Celati, C.; Lechtreck, K.F.; Mogensen, M.; Delouvée, A.; Lavia, P.; Bornens, M.; Tassin, A.-M. Part of Ran is associated with AKAP450 at the centrosome: Involvement in microtubule-organizing activity. Mol. Biol. Cell 2003, 14, 4260–4271. [Google Scholar] [CrossRef]
- Forgues, M.; Difilippantonio, M.J.; Linke, S.P.; Ried, T.; Nagashima, K.; Feden, J.; Valerie, K.; Fukasawa, K.; Wang, X.W. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol. Cell. Biol. 2003, 23, 5282–5292. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, W.; Budhu, A.; Forgues, M. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat. Cell Biol. 2005, 7, 823–830. [Google Scholar] [CrossRef]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef]
- Okuwaki, M. The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 2008, 143, 441–448. [Google Scholar] [CrossRef]
- Yang, K.; Wang, M.; Zhao, Y.; Sun, X.; Yang, Y.; Li, X.; Zhou, A.; Chu, H.; Zhou, H.; Xu, J.; et al. A redox mechanism underlying nucleolar stress sensing by nucleophosmin. Nat. Commun. 2016, 7, 13599. [Google Scholar] [CrossRef]
- Kurki, S.; Peltonen, K.; Latonen, L.; Kiviharju, T.M.; Ojala, P.M.; Meek, D.; Laiho, M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475. [Google Scholar] [CrossRef]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef]
- Sloan, K.E.; Bohnsack, K.E.; Watkins, N.J. The 5S RNP Couples p53 Homeostasis to Ribosome Biogenesis and Nucleolar Stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef]
- Colombo, E.; Marine, J.-C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef]
- Lee, S.B.; Xuan Nguyen, T.L.; Choi, J.W.; Lee, K.H.; Cho, S.W.; Liu, Z.; Ye, K.; Bae, S.S.; Ahn, J.Y. Nuclear Akt interacts with B23/NPM and protects it from proteolytic cleavage, enhancing cell survival. Proc. Natl. Acad. Sci. USA 2008, 105, 16584–16589. [Google Scholar] [CrossRef]
- Scarpa, F.J.; Paul, M.; Daringer, R.; Wolfson, W.A.; Lopez Diaz, F.; Agersborg, S.; Funari, V.A.; Weiss, L.M.; Blocker, F. TP53/NPM1-mutated acute myeloid leukemia as a molecularly distinct disease entity. J. Clin. Oncol. 2021, 39, 7030. [Google Scholar] [CrossRef]
- Li, R.; Hannon, G.J.; Beach, D.; Stillman, B. Subcellular distribution of p21 and PCNA in normal and repair-deficient cells following DNA damage. Curr. Biol. 1996, 6, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Tan, B.C.; Liu, H.; Shih, C.J.; Chien, K.Y.; Lin, C.L.; Yung, B.Y. Dephosphorylation of nucleophosmin by PP1β facilitates pRB binding and consequent E2F1-dependent DNA repair. Mol. Biol. Cell 2010, 21, 4409–4417. [Google Scholar] [CrossRef]
- Poletto, M.; Lirussi, L.; Wilson, D.M., 3rd; Tell, G. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol. Biol. Cell 2014, 25, 1641–1652. [Google Scholar] [CrossRef]
- Gibbs, E.; Perrone, B.; Hassan, A.; Kümmerle, R.; Kriwacki, R. NPM1 exhibits structural and dynamic heterogeneity upon phase separation with the p14ARF tumor suppressor. J. Magn. Reson. 2020, 310, 106646. [Google Scholar] [CrossRef]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor Suppressor ARF Degrades B23, a Nucleolar Protein Involved in Ribosome Biogenesis and Cell Proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Hwang, S.M. Classification of acute myeloid leukemia. Blood Res. 2020, 55, S1–S4. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs CHOP mutations in AML with NPM1 alteration. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef]
- SanMiguel, J.M.; Eudy, E.; Loberg, M.A.; Miles, L.A.; Stearns, T.; Mistry, J.J.; Rauh, M.J.; Levine, R.L.; Trowbridge, J.J. Cell origin-dependent cooperativity of mutant Dnmt3a and Npm1 in clonal hematopoiesis and myeloid malignancy. Blood Adv. 2022, 6, 3666–3677. [Google Scholar] [CrossRef]
- Onate, G.; Bataller, A.; Garrido, A.; Hoyos, M.; Arnan, M.; Vives, S.; Coll, R.; Tormo, M.; Sampol, A.; Escoda, L.; et al. Prognostic impact of DNMT3A mutation in acute myeloid leukemia with mutated NPM1. Blood Adv. 2022, 6, 882–890. [Google Scholar] [CrossRef]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 2031. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Haferlach, C.; Mecucci, C.; Schnittger, S.; Kohlmann, A.; Mancini, M.; Cuneo, A.; Testoni, N.; Rege-Cambrin, G.; Santucci, A.; Vignetti, M.; et al. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood 2009, 114, 3024–3032. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Alpermann, T.; Dicker, F.; Sundermann, J.; Kern, W.; Haferlach, T. Characterization of NPM1-mutated AML with a history of myelodysplastic syndromes or myeloproliferative neoplasms. Leukemia 2011, 25, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Nasillo, V.; Paolini, A.; Bettelli, F.; Pioli, V.; Giusti, D.; Gilioli, A.; Colasante, C.; Acquaviva, G.; Riva, G.; et al. NPM1-Mutated Myeloid Neoplasms with <20% Blasts: A Really Distinct Clinico-Pathologic Entity? Int. J. Mol. Sci. 2020, 21, 8975. [Google Scholar] [CrossRef]
- Itzykson, R.; Fenaux, P.; Bowen, D.; Cross, N.C.P.; Cortes, J.; De Witte, T.; Germing, U.; Onida, F.; Padron, E.; Platzbecker, U.; et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations From the European Hematology Association and the European LeukemiaNet. Hemasphere 2018, 2, e150. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Li, S.; Meloni, G.; Schnittger, S.; Gattenlohner, S.; Liso, A.; Di Ianni, M.; Martelli, M.P.; Pescarmona, E.; Foa, R.; et al. NPM1-mutated acute myeloid leukaemia occurring in JAK2-V617F+ primary myelofibrosis: De-novo origin? Leukemia 2008, 22, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Liapis, K.; Kotsianidis, I. Approaching First-Line Treatment in Patients With Advanced CMML: Hypomethylating Agents or Cytotoxic Treatment? Front. Oncol. 2021, 11, 801524. [Google Scholar] [CrossRef]
- Andraos, E.; Dignac, J.; Meggetto, F. NPM-ALK: A Driver of Lymphoma Pathogenesis and a Therapeutic Target. Cancers 2021, 13, 144. [Google Scholar] [CrossRef]
- Kuravi, S.; Baker, R.W.; Mushtaq, M.U.; Saadi, I.; Lin, T.L.; Vivian, C.J.; Valluripalli, A.; Abhyankar, S.; Ganguly, S.; Cui, W.; et al. Functional characterization of NPM1–TYK2 fusion oncogene. NPJ Precis. Oncol. 2022, 6, 3. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Bene, M.C.; et al. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Martelli, M.P. How I diagnose and treat NPM1-mutated AML. Blood 2021, 137, 589–599. [Google Scholar] [CrossRef]
- Moors, I.; Vandepoele, K.; Philippe, J.; Deeren, D.; Selleslag, D.; Breems, D.; Straetmans, N.; Kerre, T.; Denys, B. Clinical implications of measurable residual disease in AML: Review of current evidence. Crit. Rev. Oncol. Hematol. 2019, 133, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.; Potter, N.; Freeman, S.; Russell, N. How we use molecular minimal residual disease (MRD) testing in acute myeloid leukaemia (AML). Br. J. Haematol. 2021, 193, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.P.; Nibourel, O.; Berthon, C.; et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J. Clin. Oncol. 2017, 35, 185–193. [Google Scholar] [CrossRef]
- Meur, G.L.; Plesa, A.; Larcher, M.V.; Fossard, G.; Barraco, F.; Loron, S.; Balsat, M.; Ducastelle-Lepretre, S.; Gilis, L.; Thomas, X.; et al. Impact on Outcome of Minimal Residual Disease after Hematopoietic Stem Cell Transplantation with Fludarabine, Amsacrine, and Cytosine Arabinoside-Busulfan Conditioning: A Retrospective Monocentric Study. Transplant. Cell Ther. 2023, 29, 38.e31–38.e39. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gupta, A.; Kumar, S.; Mawalankar, G.; Gupta, B.; Dhole, N.; Kori, R.; Singh, A. Flow cytometric measurable residual disease in adult acute myeloid leukemia: A preliminary report from Eastern India. J. Hematop. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Kern, W.; Haferlach, C.; Bacher, U.; Haferlach, T.; Schnittger, S. Flow cytometric identification of acute myeloid leukemia with limited differentiation and NPM1 type A mutation: A new biologically defined entity. Leukemia 2009, 23, 1361–1364. [Google Scholar] [CrossRef]
- Pettersson, L.; Johansson Alm, S.; Almstedt, A.; Chen, Y.; Orrsjo, G.; Shah-Barkhordar, G.; Zhou, L.; Kotarsky, H.; Vidovic, K.; Asp, J.; et al. Comparison of RNA- and DNA-based methods for measurable residual disease analysis in NPM1-mutated acute myeloid leukemia. Int. J. Lab. Hematol. 2021, 43, 664–674. [Google Scholar] [CrossRef]
- Jennings, L.J. Normalization of NPM1 mutant transcript to the wild-type transcript. eJHaem 2022, 3, 1343–1345. [Google Scholar] [CrossRef]
- Quiros, P.M.; Gu, M.; Barcena, C.; Iyer, V.; Vassiliou, G.S. NPM1 gene mutations can be confidently identified in blood DNA months before de novo AML onset. Blood Adv. 2022, 6, 2409–2413. [Google Scholar] [CrossRef]
- Lesieur, A.; Thomas, X.; Nibourel, O.; Boissel, N.; Fenwarth, L.; De Botton, S.; Fournier, E.; Celli-Lebras, K.; Raffoux, E.; Recher, C.; et al. Minimal residual disease monitoring in acute myeloid leukemia with non-A/B/D-NPM1 mutations by digital polymerase chain reaction: Feasibility and clinical use. Haematologica 2021, 106, 1767–1769. [Google Scholar] [CrossRef]
- Koh, Y.; Park, J.; Bae, E.K.; Ahn, K.S.; Kim, I.; Bang, S.M.; Lee, J.H.; Yoon, S.S.; Lee, D.S.; Lee, Y.Y.; et al. Non-A type nucleophosmin 1 gene mutation predicts poor clinical outcome in de novo adult acute myeloid leukemia: Differential clinical importance of NPM1 mutation according to subtype. Int. J. Hematol. 2009, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.M.; et al. Molecular subtypes of NPM1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, E55–E58. [Google Scholar] [CrossRef]
- Ediriwickrema, A.; Aleshin, A.; Reiter, J.G.; Corces, M.R.; Kohnke, T.; Stafford, M.; Liedtke, M.; Medeiros, B.C.; Majeti, R. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020, 4, 943–952. [Google Scholar] [CrossRef]
- Li, K.; Du, Y.; Cai, Y.; Liu, W.; Lv, Y.; Huang, B.; Zhang, L.; Wang, Z.; Liu, P.; Sun, Q.; et al. Single-cell analysis reveals the chemotherapy-induced cellular reprogramming and novel therapeutic targets in relapsed/refractory acute myeloid leukemia. Leukemia 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Duchmann, M.; Joudinaud, R.; Boudry, A.; Pasanisi, J.; Di Feo, G.; Kim, R.; Bucci, M.; Chauvel, C.; Chat, L.; Larcher, L.; et al. Hematopoietic differentiation at single-cell resolution in NPM1-mutated AML. Blood Cancer J. 2022, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Tislevoll, B.S.; Hellesoy, M.; Fagerholt, O.H.E.; Gullaksen, S.E.; Srivastava, A.; Birkeland, E.; Kleftogiannis, D.; Ayuda-Duran, P.; Piechaczyk, L.; Tadele, D.S.; et al. Early response evaluation by single cell signaling profiling in acute myeloid leukemia. Nat. Commun. 2023, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Dohner, K.; Krauter, J.; Frohling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Spath, D.; Morgan, M.; Benner, A.; et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.E.; Lamb, K.; Allen, C.; El-Sharkawi, D.; Stowe, C.; Jenkinson, S.; Tinsley, S.; Dickson, G.; Burnett, A.K.; Hills, R.K.; et al. Simpson’s Paradox and the Impact of Different DNMT3A Mutations on Outcome in Younger Adults With Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, P.; Renneville, A.; Ben Abdelali, R.; Nibourel, O.; Thomas, X.; Pautas, C.; de Botton, S.; Raffoux, E.; Cayuela, J.M.; Boissel, N.; et al. Impact of additional genetic alterations on the outcome of patients with NPM1-mutated cytogenetically normal acute myeloid leukemia. Haematologica 2015, 100, e196–e199. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Ivey, A.; Grimwade, D.; Group, U.K.N.C.R.I.A.W. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 375, e9. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.; Hills, R.; Freeman, S.; Potter, N.; Jovanovic, J.; Ivey, A.; Kanda, A.S.; Runglall, M.; Foot, N.; Valganon, M.; et al. Molecular MRD status and outcome after transplantation in NPM1-mutated AML. Blood 2020, 135, 680–688. [Google Scholar] [CrossRef]
- Bill, M.; Grimm, J.; Jentzsch, M.; Kloss, L.; Goldmann, K.; Schulz, J.; Beinicke, S.; Hantschel, J.; Cross, M.; Vucinic, V.; et al. Digital droplet PCR-based absolute quantification of pre-transplant NPM1 mutation burden predicts relapse in acute myeloid leukemia patients. Ann. Hematol. 2018, 97, 1757–1765. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; Bixby, D.L.; et al. CPX-351 versus 7+3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2021, 8, e481–e491. [Google Scholar] [CrossRef]
- Nadas, J.; Sun, D. Anthracyclines as effective anticancer drugs. Expert Opin. Drug Discov. 2006, 1, 549–568. [Google Scholar] [CrossRef]
- Neuendorff, N.R.; Loh, K.P.; Mims, A.S.; Christofyllakis, K.; Soo, W.K.; Bölükbasi, B.; Oñoro-Algar, C.; Hundley, W.G.; Klepin, H.D. Anthracycline-related cardiotoxicity in older patients with acute myeloid leukemia: A Young SIOG review paper. Blood Adv. 2020, 4, 762–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beretta, G.L.; Zunino, F. Molecular Mechanisms of Anthracycline Activity. In Anthracycline Chemistry and Biology II: Mode of Action, Clinical Aspects and New Drugs; Krohn, K., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2008; pp. 1–19. [Google Scholar]
- Wang, A.H.; Ughetto, G.; Quigley, G.J.; Rich, A. Interactions between an anthracycline antibiotic and DNA: Molecular structure of daunomycin complexed to d(CpGpTpApCpG) at 1.2-A resolution. Biochemistry 1987, 26, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [PubMed]
- van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021, 288, 6095–6111. [Google Scholar] [CrossRef]
- Morimoto, S.; Tsuda, M.; Bunch, H.; Sasanuma, H.; Austin, C.; Takeda, S. Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes 2019, 10, 868. [Google Scholar] [CrossRef]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef]
- Tamura, T.; Ishihara, M.; Lamphier, M.S.; Tanaka, N.; Oishi, I.; Aizawa, S.; Matsuyama, T.; Mak, T.W.; Taki, S.; Taniguchi, T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature 1995, 376, 596–599. [Google Scholar] [CrossRef]
- Doyle, S.E.; Vaidya, S.A.; O’Connell, R.; Dadgostar, H.; Dempsey, P.W.; Wu, T.-T.; Rao, G.; Sun, R.; Haberland, M.E.; Modlin, R.L.; et al. IRF3 Mediates a TLR3/TLR4-Specific Antiviral Gene Program. Immunity 2002, 17, 251–263. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Malfatti, M.C.; Gerratana, L.; Dalla, E.; Isola, M.; Damante, G.; Di Loreto, C.; Puglisi, F.; Tell, G. APE1 and NPM1 protect cancer cells from platinum compounds cytotoxicity and their expression pattern has a prognostic value in TNBC. J. Exp. Clin. Cancer Res. 2019, 38, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rechkoblit, O.; Johnson, R.E.; Buku, A.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural insights into mutagenicity of anticancer nucleoside analog cytarabine during replication by DNA polymerase η. Sci. Rep. 2019, 9, 16400. [Google Scholar] [CrossRef] [PubMed]
- Prakasha Gowda, A.S.; Polizzi, J.M.; Eckert, K.A.; Spratt, T.E. Incorporation of gemcitabine and cytarabine into DNA by DNA polymerase beta and ligase III/XRCC1. Biochemistry 2010, 49, 4833–4840. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Döhner, K.; Schlenk, R.F.; Habdank, M.; Scholl, C.; Rücker, F.G.; Corbacioglu, A.; Bullinger, L.; Fröhling, S.; Döhner, H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: Interaction with other gene mutations. Blood 2005, 106, 3740–3746. [Google Scholar] [CrossRef]
- Pastore, F.; Greif, P.A.; Schneider, S.; Ksienzyk, B.; Mellert, G.; Zellmeier, E.; Braess, J.; Sauerland, C.M.; Heinecke, A.; Krug, U.; et al. The NPM1 mutation type has no impact on survival in cytogenetically normal AML. PLoS ONE 2014, 9, e109759. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Propper, D.J.; McDonald, A.C.; Man, A.; Thavasu, P.; Balkwill, F.; Braybrooke, J.P.; Caponigro, F.; Graf, P.; Dutreix, C.; Blackie, R.; et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J. Clin. Oncol. 2001, 19, 1485–1492. [Google Scholar] [CrossRef]
- Stölzel, F.; Steudel, C.; Oelschlägel, U.; Mohr, B.; Koch, S.; Ehninger, G.; Thiede, C. Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann. Hematol. 2010, 89, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Müller, C.; Grüning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Lazarus, H.M. Midostaurin: An emerging treatment for acute myeloid leukemia patients. J. Blood Med. 2016, 7, 73–83. [Google Scholar] [CrossRef]
- Juliusson, G.; Jädersten, M.; Deneberg, S.; Lehmann, S.; Möllgård, L.; Wennström, L.; Antunovic, P.; Cammenga, J.; Lorenz, F.; Ölander, E.; et al. The prognostic impact of FLT3-ITD and NPM1 mutation in adult AML is age-dependent in the population-based setting. Blood Adv. 2020, 4, 1094–1101. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.R.; McCloskey, J.; Smith, C.C.; Schiller, G.J.; Bradley, T.; et al. Venetoclax in Combination with Gilteritinib Demonstrates Molecular Clearance of FLT3 mutation in Relapsed/Refractory FLT3-Mutated Acute Myeloid Leukemia. Blood 2021, 138, 691. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Russell, N. Twenty five years of UK trials in acute myeloid leukaemia: What have we learned? Br. J. Haematol. 2020, 188, 86–100. [Google Scholar] [CrossRef]
- De Propris, M.S.; Raponi, S.; Diverio, D.; Milani, M.L.; Meloni, G.; Falini, B.; Foà, R.; Guarini, A. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica 2011, 96, 1548–1551. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.L.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Paschka, P.; Krzykalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T.; et al. Gemtuzumab Ozogamicin in NPM1-Mutated Acute Myeloid Leukemia: Early Results From the Prospective Randomized AMLSG 09-09 Phase III Study. J. Clin. Oncol. 2020, 38, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D. Antibody-Drug Conjugates of Calicheamicin Derivative: Gemtuzumab Ozogamicin and Inotuzumab Ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef] [PubMed]
- Herbener, P.; Schönfeld, K.; König, M.; Germer, M.; Przyborski, J.M.; Bernöster, K.; Schüttrumpf, J. Functional relevance of in vivo half antibody exchange of an IgG4 therapeutic antibody-drug conjugate. PLoS ONE 2018, 13, e0195823. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- Lama, D.; Sankararamakrishnan, R. Identification of core structural residues in the sequentially diverse and structurally homologous Bcl-2 family of proteins. Biochemistry 2010, 49, 2574–2584. [Google Scholar] [CrossRef]
- Du, H.; Wolf, J.; Schafer, B.; Moldoveanu, T.; Chipuk, J.E.; Kuwana, T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011, 286, 491–501. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Zhang, S.; Qin, F.; Yang, L.; Xian, J.; Zou, Q.; Jin, H.; Wang, L.; Zhang, L. Nucleophosmin Mutations Induce Chemosensitivity in THP-1 Leukemia Cells by Suppressing NF-κB Activity and Regulating Bax/Bcl-2 Expression. J. Cancer 2016, 7, 2270–2279. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Reville, P.K.; Kantarjian, H.; Jabbour, E.; Borthakur, G.; Daver, N.; Issa, G.; Furudate, K.; Tanaka, T.; Pierce, S.; et al. Contemporary outcomes in IDH-mutated acute myeloid leukemia: The impact of co-occurring NPM1 mutations and venetoclax-based treatment. Am. J. Hematol. 2022, 97, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Kon Kim, T.; Gore, S.D.; Zeidan, A.M. Epigenetic Therapy in Acute Myeloid Leukemia: Current and Future Directions. Semin. Hematol. 2015, 52, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Roboz, G.J.; Wei, A.H.; Döhner, H.; Pocock, C.; Selleslag, D.; Montesinos, P.; Sayar, H.; Musso, M.; Figuera-Alvarez, A.; et al. Management of adverse events in patients with acute myeloid leukemia in remission receiving oral azacitidine: Experience from the phase 3 randomized QUAZAR AML-001 trial. J. Hematol. Oncol. 2021, 14, 133. [Google Scholar] [CrossRef]
- Wu, H.C.; Rérolle, D.; Berthier, C.; Hleihel, R.; Sakamoto, T.; Quentin, S.; Benhenda, S.; Morganti, C.; Wu, C.; Conte, L.; et al. Actinomycin D Targets NPM1c-Primed Mitochondria to Restore PML-Driven Senescence in AML Therapy. Cancer Discov. 2021, 11, 3198–3213. [Google Scholar] [CrossRef]
- Gionfriddo, I.; Brunetti, L.; Mezzasoma, F.; Milano, F.; Cardinali, V.; Ranieri, R.; Venanzi, A.; Pierangeli, S.; Vetro, C.; Spinozzi, G.; et al. Dactinomycin induces complete remission associated with nucleolar stress response in relapsed/refractory NPM1-mutated AML. Leukemia 2021, 35, 2552–2562. [Google Scholar] [CrossRef]
- Cho, H.; Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Chung, H.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. Arsenic trioxide synergistically promotes the antileukaemic activity of venetoclax by downregulating Mcl-1 in acute myeloid leukaemia cells. Exp. Hematol. Oncol. 2021, 10, 28. [Google Scholar] [CrossRef]
- Chin, L.; Kumana, C.; Kwong, Y.-L.; Gill, H. The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics 2022, 14, 1945. [Google Scholar] [CrossRef]
- El-Hajj, H.; Dassouki, Z.; Berthier, C.; Raffoux, E.; Ades, L.; Legrand, O.; Hleihel, R.; Sahin, U.; Tawil, N.; Salameh, A.; et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM-1 resulting in apoptosis of AML cells. Blood 2015, 125, 3447–3454. [Google Scholar] [CrossRef]
- Huang, M.; Thomas, D.; Li, M.; Feng, W.; Chan, S.; Majeti, R.; Mitchell, B. Role of cysteine 288 in nucleophosmin cytoplasmic mutations: Sensitization to toxicity induced by arsenic trioxide and bortezomib. Leukemia 2013, 27, 1970–1980. [Google Scholar] [CrossRef]
- Pianigiani, G.; Gagliardi, A.; Mezzasoma, F.; Rocchio, F.; Tini, V.; Bigerna, B.; Sportoletti, P.; Caruso, S.; Marra, A.; Peruzzi, S.; et al. Prolonged XPO1 inhibition is essential for optimal antileukemic activity in NPM1-mutated AML. Blood Adv. 2022, 6, 5938–5949. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.A.; Friedlander, S.Y.; Arrate, M.P.; Chang, H.; Gorska, A.E.; Fuller, L.D.; Ramsey, H.E.; Kashyap, T.; Argueta, C.; Debler, S.; et al. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv. 2020, 4, 586–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cierpicki, T.; Grembecka, J. Challenges and opportunities in targeting the menin-MLL interaction. Future Med. Chem. 2014, 6, 447–462. [Google Scholar] [CrossRef]
- Fiskus, W.; Boettcher, S.; Daver, N.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Takahashi, K.; Kadia, T.M.; DiNardo, C.D.; et al. Effective Menin inhibitor-based combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J. 2022, 12, 5. [Google Scholar] [CrossRef]
- Uckelmann, H.J.; Haarer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; Perner, F.; Rajput, M.; Antonissen, N.J.C.; Wen, Y.; et al. Mutant NPM1 Directly Regulates Oncogenic Transcription in Acute Myeloid Leukemia. Cancer Discov. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Ghasemi, R.; Struthers, H.; Wilson, E.R.; Spencer, D.H. Contribution of CTCF binding to transcriptional activity at the HOXA locus in NPM1-mutant AML cells. Leukemia 2021, 35, 404–416. [Google Scholar] [CrossRef]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.A.; Wang, E.S. Menin Inhibitors in Acute Myeloid Leukemia-What Does the Future Hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Riva, G.; Lagreca, I.; Barozzi, P.; Bettelli, F.; Paolini, A.; Nasillo, V.; Lusenti, B.; Pioli, V.; Giusti, D.; et al. Neoantigen-Specific T-Cell Immune Responses: The Paradigm of NPM1-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 9159. [Google Scholar] [CrossRef]
- Kuzelova, K.; Brodska, B.; Schetelig, J.; Röllig, C.; Ráčil, Z.; Stickel, J.; Helbig, G.; Fuchs, O.; Vraná, M.; Pecherkova, P.; et al. Association of HLA class I type with prevalence and outcome of patients with acute myeloid leukemia and mutated nucleophosmin. PLoS ONE 2018, 13, e0204290. [Google Scholar] [CrossRef]
- Dong, H.; Ham, J.D.; Hu, G.; Xie, G.; Vergara, J.; Liang, Y.; Ali, A.; Tarannum, M.; Donner, H.; Baginska, J.; et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2022, 119, e2122379119. [Google Scholar] [CrossRef]
- van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.; Kester, M.G.; van der Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M.; et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Investig. 2019, 129, 774–785. [Google Scholar] [CrossRef]
- Perriello, V.M.; Rotiroti, M.C.; Pisani, I.; Alberti, G.; Pianigiani, G.; Rossi, R.; Ciaurro, V.; Serafini, M.; Martelli, M.P.; Falini, B. CD123 and CD33 Co-Targeting By Balanced Signaling on CAR-CIK Cells Reduces Potential Off-Target Toxicity While Preserving the Anti-Leukemic Activity of Acute Myeloid Leukemia. Blood 2021, 138, 1699. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.M.; Schneider, V.; Schrezenmeier, H.; Hofmann, S.; Götz, M. Enhanced Stimulation of Antigen-Specific Immune Responses Against NPM1-Mutated AML. Blood 2021, 138, 1292. [Google Scholar] [CrossRef]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef]
- Currie, K.S.; Kropf, J.E.; Lee, T.; Blomgren, P.; Xu, J.; Zhao, Z.; Gallion, S.; Whitney, J.A.; Maclin, D.; Lansdon, E.B.; et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J. Med. Chem. 2014, 57, 3856–3873. [Google Scholar] [CrossRef]
- Walker, A.R.; Byrd, J.C.; Blachly, J.S.; Bhatnagar, B.; Mims, A.S.; Orwick, S.; Lin, T.L.; Crosswell, H.E.; Zhang, D.; Minden, M.D.; et al. Entospletinib in Combination with Induction Chemotherapy in Previously Untreated Acute Myeloid Leukemia: Response and Predictive Significance of HOXA9 and MEIS1 Expression. Clin. Cancer Res. 2020, 26, 5852–5859. [Google Scholar] [CrossRef]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Rönnstrand, L.; et al. SYK Is a Critical Regulator of FLT3 in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef]
- Liu, N.; Wu, Y.; Wen, X.; Li, P.; Lu, F.; Shang, H. Chronic stress promotes acute myeloid leukemia progression through HMGB1/NLRP3/IL-1β signaling pathway. J. Mol. Med. 2021, 99, 403–414. [Google Scholar] [CrossRef]
- Sakhnevych, S.S.; Yasinska, I.M.; Bratt, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Hussain, R.; Siligardi, G.; Fiedler, W.; Wellbrock, J.; Gibbs, B.F.; et al. Cortisol facilitates the immune escape of human acute myeloid leukemia cells by inducing latrophilin 1 expression. Cell. Mol. Immunol. 2018, 15, 994–997. [Google Scholar] [CrossRef]
- Eckerling, A.; Ricon-Becker, I.; Sorski, L.; Sandbank, E.; Ben-Eliyahu, S. Stress and cancer: Mechanisms, significance and future directions. Nat. Rev. Cancer 2021, 21, 767–785. [Google Scholar] [CrossRef]
- Fucà, G.; Galli, G.; Poggi, M.; Lo Russo, G.; Proto, C.; Imbimbo, M.; Ferrara, R.; Zilembo, N.; Ganzinelli, M.; Sica, A.; et al. Modulation of peripheral blood immune cells by early use of steroids and its association with clinical outcomes in patients with metastatic non-small cell lung cancer treated with immune checkpoint inhibitors. ESMO Open 2019, 4, e000457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.J.; Atenafu, E.G.; Schimmer, A.D.; Minden, M.D.; Chang, H. Expression of CD4 is correlated with an unfavorable prognosis in wild-type NPM1, FLT3-ITD-negative cytogenetically normal adult acute myeloid leukemia. Int. J. Lab. Hematol. 2017, 39, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Jiang, H.; Brandwein, J.; Kamel-Reid, S.; Chang, H. Prognostic factors in normal karyotype acute myeloid leukemia in the absence of the FLT3-ITD mutation. Leuk. Res. 2011, 35, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Bermúdez, M.-L.; Koka, M.; Chandran, B.; Pawale, D.; Vishnubhotla, R.; Alankar, S.; Maturi, R.; Subramaniam, B.; Sadhasivam, S. Large-scale genomic study reveals robust activation of the immune system following advanced Inner Engineering meditation retreat. Proc. Natl. Acad. Sci. USA 2021, 118, e2110455118. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chin, L.; Wong, C.Y.G.; Gill, H. Targeting and Monitoring Acute Myeloid Leukaemia with Nucleophosmin-1 (NPM1) Mutation. Int. J. Mol. Sci. 2023, 24, 3161. https://doi.org/10.3390/ijms24043161
Chin L, Wong CYG, Gill H. Targeting and Monitoring Acute Myeloid Leukaemia with Nucleophosmin-1 (NPM1) Mutation. International Journal of Molecular Sciences. 2023; 24(4):3161. https://doi.org/10.3390/ijms24043161
Chicago/Turabian StyleChin, Lynn, Chantelle Ye Gwen Wong, and Harinder Gill. 2023. "Targeting and Monitoring Acute Myeloid Leukaemia with Nucleophosmin-1 (NPM1) Mutation" International Journal of Molecular Sciences 24, no. 4: 3161. https://doi.org/10.3390/ijms24043161