

The Chromosome-Level Genome of Hestina assimilis (Lepidoptera: Nymphalidae) Reveals the Evolution of Saprophagy-Related Genes in Brush-Footed Butterflies
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. High-Quality Chromosome-Level Assembly, Gene Prediction, and Functional Annotation
2.2. Synteny Analysis
2.3. Phylogenetic and Positive Selection Analysis
2.4. Gene Family Associated with Diet Metabolism Analysis
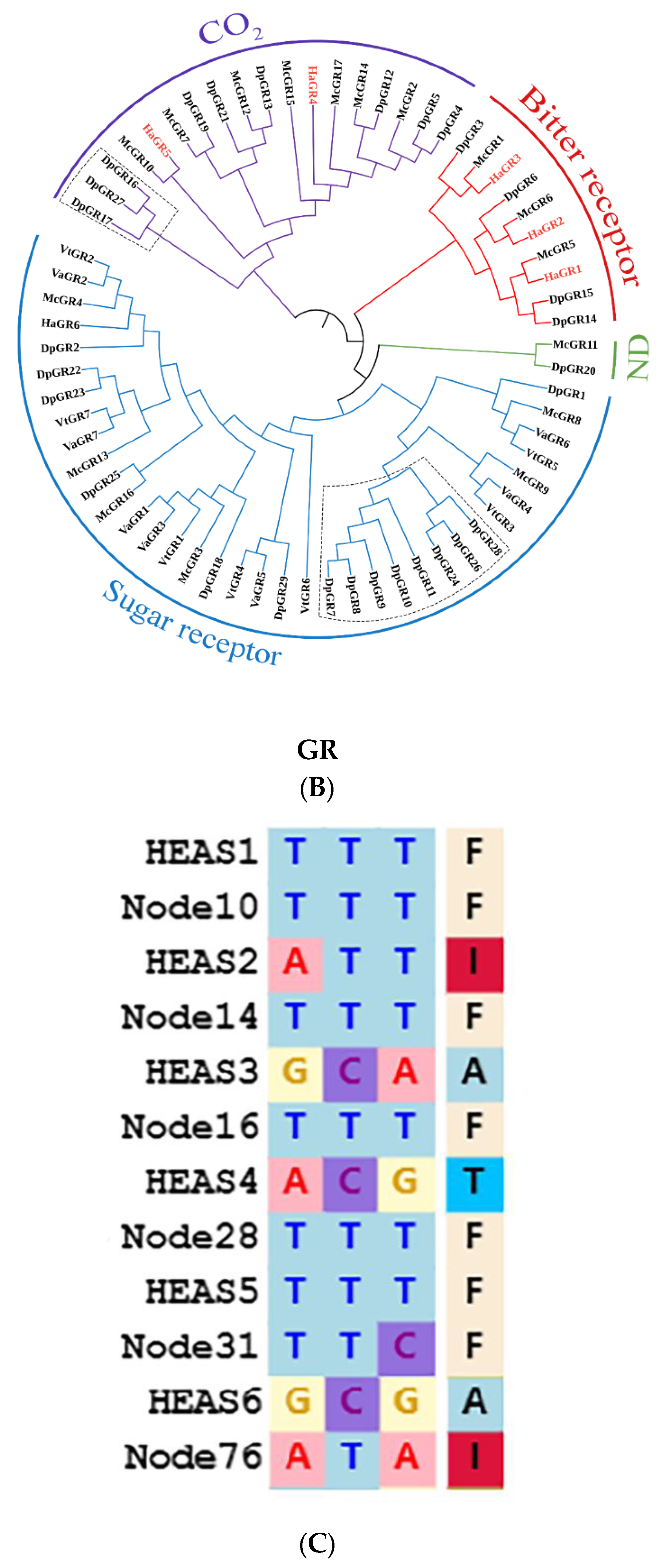
2.5. Evolution of P450, OBP, OR, and GR Gene Families Related to Saprophagy in H. assimilis
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Sequencing
4.2. Genome Assembly and Assessment
4.3. Repetitive Elements, Gene Prediction, and Gene Functional Annotation
4.4. Phylogenetic Reconstruction and Gene Family Analyses
4.5. Positive Selection Analysis
4.6. The Evolution of P450, OBP, OR, and GR Gene Families Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hunter, J.P. Key innovations and the ecology of macroevolution. Trends Ecol. Evol. 1998, 13, 31–36. [Google Scholar] [CrossRef]
- Geister, T.L.; Lorenz, M.W.; Hoffmann, K.H.; Fischer, K. Adult nutrition and butterfly fitness: Effects of diet quality on reproductive output, egg composition, and egg hatching success. Front. Zool. 2008, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Guerrero, Y.T.; Ibarra-Laclette, E.; Martínez Del Río, C.; Barrera-Redondo, J.; Rebollar, E.A.; Ortega, J.; León-Paniagua, L.; Urrutia, A.; Aguirre-Planter, E.; Eguiarte, L.E. Genomic consequences of dietary diversification and parallel evolution due to nectarivory in leaf-nosed bats. GigaScience 2020, 9, giaa059. [Google Scholar] [CrossRef]
- Lane, K.A.; Lucas, K.M.; Yack, J.E. Hearing in a diurnal, mute butterfly, Morpho peleides (Papilionoidea, Nymphalidae). J. Comp. Neurol. 2008, 508, 677–686. [Google Scholar] [CrossRef]
- Omura, H.; Honda, K. Feeding responses of adult butterflies, Nymphalis xanthomelas, Kaniska canace and Vanessa indica, to components in tree sap and rotting fruits: Synergistic effects of ethanol and acetic acid on sugar responsiveness. J. Insect Physiol. 2003, 49, 1031–1038. [Google Scholar] [CrossRef]
- Omura, H. Identification of feeding attractants in oak sap for adults of two nymphalid butterflies, Kniska canace and Vanessa indica (vol 25, pg 281, 2000). Physiol. Entomol. 2001, 26, 283. [Google Scholar]
- Zhou, Y.N.; Xie, S.; Chen, J.N.; Wang, Z.H.; Yang, P.; Zhou, S.C.; Pang, L.; Li, F.; Shi, M.; Huang, J.H.; et al. Expression and functional characterization of odorant-binding protein genes in the endoparasitic wasp Cotesia vestalis. Insect Sci. 2021, 28, 1354–1368. [Google Scholar] [CrossRef]
- Hill, J.; Hamer, K.; Tangah, J.; Dawood, M. Ecology of tropical butterflies in rainforest gaps. Oecologia 2001, 128, 294–302. [Google Scholar] [CrossRef]
- Omura, H.; Honda, K.; Asaoka, K.; Inoue, T.A. Tolerance to fermentation products in sugar reception: Gustatory adaptation of adult butterfly proboscis for feeding on rotting foods. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2008, 194, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Kaliszewska, Z.A.; Lohman, D.J.; Sommer, K.; Adelson, G.; Rand, D.B.; Mathew, J.; Talavera, G.; Pierce, N.E. When caterpillars attack: Biogeography and life history evolution of the Miletinae (Lepidoptera: Lycaenidae). Evol. Int. J. Org. Evol. 2015, 69, 571–588. [Google Scholar] [CrossRef]
- Li, Z.H.; Hua, C.; Wei-Yan, Y.U.; Song, D.J.; Chen, H.; Shan-Jin, X.U.; Tang, F. Bionomics of the butterfly, Hestina assimilis. Chin. J. Entomol. 2008, 45, 754–757. [Google Scholar]
- Au, T.F.; Bonebrake, T.C. Increased Suitability of Poleward Climate for a Tropical Butterfly (Euripus nyctelius) (Lepidoptera: Nymphalidae) Accompanies its Successful Range Expansion. J. Insect Sci. 2019, 19, 2. [Google Scholar] [CrossRef]
- Tanio, T.; Kuramoto, N. Effects of temperature on the initiation of post-diapause development in larvae of Hestina assimilis assimilis (Lepidoptera, Nymphalidae) and Hestina persimilis japonica. Lepid. Sci. 2019, 70, 59–63. [Google Scholar]
- Zhang, S.; Zhang, Z.; Wang, H.; Kong, X. Antennal transcriptome analysis and comparison of olfactory genes in two sympatric defoliators, Dendrolimus houi and Dendrolimus kikuchii (Lepidoptera: Lasiocampidae). Insect Biochem. Mol. Biol. 2014, 52, 69–81. [Google Scholar] [CrossRef]
- Konorov, E.A.; Nikitin, M.A.; Mikhailov, K.V.; Lysenkov, S.N.; Belenky, M.; Chang, P.L.; Nuzhdin, S.V.; Scobeyeva, V.A. Genomic exaptation enables Lasius niger adaptation to urban environments. BMC Evol. Biol. 2017, 17, 39. [Google Scholar] [CrossRef] [Green Version]
- Indra, R.; Moserova, M.; Sulc, M.; Frei, E.; Stiborova, M. Oxidation of carcinogenic benzo[a]pyrene by human and rat cytochrome P450 1A1 and its influencing by cytochrome b5-a comparative study. Neuro Endocrinol. Lett. 2013, 34 (Suppl. 2), 55–63. [Google Scholar]
- Wen, Z.; Baudry, J.; Berenbaum, M.R.; Schuler, M.A. Ile115Leu mutation in the SRS1 region of an insect cytochrome P450 (CYP6B1) compromises substrate turnover via changes in a predicted product release channel. Protein Eng. Des. Sel. 2005, 18, 191–199. [Google Scholar] [CrossRef]
- Li, X.; Baudry, J.; Berenbaum, M.R.; Schuler, M.A. Structural and functional divergence of insect CYP6B proteins: From specialist to generalist cytochrome P450. Proc. Natl. Acad. Sci. USA 2004, 101, 2939–2944. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Shi, Q.; Li, Q.; Wang, R.; Xu, C.; Wang, H.; Ran, C.; Song, Y.; Zeng, R. Identification of a cytochrome P450 CYP6AB60 gene associated with tolerance to multi-plant allelochemicals from a polyphagous caterpillar tobacco cutworm (Spodoptera litura). Pestic. Biochem. Physiol. 2019, 154, 60–66. [Google Scholar] [CrossRef]
- Fu, N.; Li, J.; Ren, L.; Li, X.; Wang, M.; Li, F.; Zong, S.; Luo, Y. Chromosome-level genome assembly of Monochamus saltuarius reveals its adaptation and interaction mechanism with pine wood nematode. Int. J. Biol. Macromol. 2022, 222, 325–336. [Google Scholar] [CrossRef]
- Feyereisen, R. Evolution of insect P450. Biochem. Soc. Trans. 2006, 34, 1252–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhu, S.; Jia, Q.; Yuan, D.; Ren, C.; Li, K.; Liu, S.; Cui, Y.; Zhao, H.; Cao, Y.; et al. The genomic and functional landscapes of developmental plasticity in the American cockroach. Nat. Commun. 2018, 9, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, T.D. Proteins and peptides as pheromone signals and chemical signatures. Anim. Behav. 2014, 97, 273–280. [Google Scholar] [CrossRef]
- Himmel, N.J.; Benton, R. Sweet sensors support stressed cell survival. PLoS Biol. 2022, 20, e3001705. [Google Scholar] [CrossRef]
- Herness, M.S.; Gilbertson, T.A. Cellular mechanisms of taste transduction. Annu. Rev. Physiol. 1999, 61, 873–900. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Reilly, P.F.; Lewis, J.J.; Reed, R.D.; Andolfatto, P.; Walters, J.R. Dichotomy of Dosage Compensation along the Neo Z Chromosome of the Monarch Butterfly. Curr. Biol. CB 2019, 29, 4071–4077.e4073. [Google Scholar] [CrossRef] [Green Version]
- Mongue, A.J.; Nguyen, P.; Voleníková, A.; Walters, J.R. Neo-sex Chromosomes in the Monarch Butterfly, Danaus plexippus. G3 Genes|Genomes|Genetics 2017, 7, 3281–3294. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, Y.X.; Wang, W.L.; Cao, Y.; Li, S.; Zhang, B.X.; Li, S.G. Identification of putative cytochrome P450 monooxygenase genes from the small white butterfly, Pieris rapae (Lepidoptera: Pieridae), and their response to insecticides. Arch. Insect Biochem. Physiol. 2018, 98, e21455. [Google Scholar] [CrossRef]
- Zhao, P.; Xue, H.; Zhu, X.; Wang, L.; Zhang, K.; Li, D.; Ji, J.; Niu, L.; Gao, X.; Luo, J.; et al. Silencing of cytochrome P450 gene CYP321A1 effects tannin detoxification and metabolism in Spodoptera litura. Int. J. Biol. Macromol. 2022, 194, 895–902. [Google Scholar] [CrossRef]
- Feyereisen, R. Origin and evolution of the CYP4G subfamily in insects, cytochrome P450 enzymes involved in cuticular hydrocarbon synthesis. Mol. Phylogenetics Evol. 2020, 143, 106695. [Google Scholar] [CrossRef]
- Niimura, Y.; Matsui, A.; Touhara, K. Acceleration of Olfactory Receptor Gene Loss in Primate Evolution: Possible Link to Anatomical Change in Sensory Systems and Dietary Transition. Mol. Biol. Evol. 2018, 35, 1437–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Wang, X.Y.; Tan, L.S.; Lu, W.; Zheng, X.L. Identification of Chemosensory Genes, Including Candidate Pheromone Receptors, in Phauda flammans (Walker) (Lepidoptera: Phaudidae) Through Transcriptomic Analyses. Front. Physiol. 2022, 13, 907694. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.; Guerenstein, P.G.; Mechaber, W.L.; Hildebrand, J.G. Floral CO2 reveals flower profitability to moths. J. Chem. Ecol. 2004, 30, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Papanicolaou, A.; Zhang, H.J.; Anderson, A. Expansion of a bitter taste receptor family in a polyphagous insect herbivore. Sci. Rep. 2016, 6, 23666. [Google Scholar] [CrossRef]
- De Coster, W.; De Rijk, P.; De Roeck, A.; De Pooter, T.; D’Hert, S.; Strazisar, M.; Sleegers, K.; Van Broeckhoven, C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [Green Version]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Guiglielmoni, N.; Houtain, A.; Derzelle, A.; Van Doninck, K.; Flot, J.F. Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinform. 2021, 22, 303. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, S.; Zhao, Q.; Ming, R.; Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 2019, 5, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. Using BUSCO to Assess Insect Genomic Resources. Methods Mol. Biol. 2019, 1858, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-Genome Annotation with BRAKER. Methods Mol. Biol. 2019, 1962, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Mulder, N.; Apweiler, R. InterPro and InterProScan: Tools for protein sequence classification and comparison. Methods Mol. Biol. (Clifton N.J.) 2007, 396, 59–70. [Google Scholar] [CrossRef]
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007, 2007, pdb.top17. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazot, N.; Wahlberg, N.; Freitas, A.V.L.; Mitter, C.; Labandeira, C.; Sohn, J.C.; Sahoo, R.K.; Seraphim, N.; de Jong, R.; Heikkilä, M. Priors and Posteriors in Bayesian Timing of Divergence Analyses: The Age of Butterflies Revisited. Syst. Biol. 2019, 68, 797–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, A.Y.; Plotkin, D.; Espeland, M.; Meusemann, K.; Toussaint, E.F.A.; Donath, A.; Gimnich, F.; Frandsen, P.B.; Zwick, A.; Dos Reis, M.; et al. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl. Acad. Sci. USA 2019, 116, 22657–22663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. CABIOS 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Cui, Y.; Ren, Y.D.; Lyu, M.; Zheng, S.C.; Feng, Q.L.; Xiang, H. Genomic divergences between the two polyphagous Spodoptera relatives provide cues for successful invasion of the fall armyworm. Insect Sci. 2020, 27, 1257–1265. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Clan | H. assimilis | M. cinxia | V. tameamea | V. atalanta | D. plexippus |
|---|---|---|---|---|---|---|
| P450 | Clan 3 | 23 | 36 | 28 | 30 | 33 |
| Clan 4 | 39 | 23 | 20 | 32 | 23 | |
| Clan 2 | 11 | 9 | 8 | 7 | 13 | |
| Mitochondrial | 7 | 10 | 11 | 18 | 9 | |
| OR | 33 | 43 | 43 | 52 | 57 | |
| OBP | 20 | 25 | 25 | 29 | 32 | |
| GR | 6 | 17 | 7 | 7 | 29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Li, X.-D.; Jiang, T.; Wang, H.; Dan, Z.; Xu, S.-Q.; Guan, D.-L. The Chromosome-Level Genome of Hestina assimilis (Lepidoptera: Nymphalidae) Reveals the Evolution of Saprophagy-Related Genes in Brush-Footed Butterflies. Int. J. Mol. Sci. 2023, 24, 2087. https://doi.org/10.3390/ijms24032087
Zhao L, Li X-D, Jiang T, Wang H, Dan Z, Xu S-Q, Guan D-L. The Chromosome-Level Genome of Hestina assimilis (Lepidoptera: Nymphalidae) Reveals the Evolution of Saprophagy-Related Genes in Brush-Footed Butterflies. International Journal of Molecular Sciences. 2023; 24(3):2087. https://doi.org/10.3390/ijms24032087
Chicago/Turabian StyleZhao, Lu, Xiao-Dong Li, Tao Jiang, Hang Wang, Zhicuo Dan, Sheng-Quan Xu, and De-Long Guan. 2023. "The Chromosome-Level Genome of Hestina assimilis (Lepidoptera: Nymphalidae) Reveals the Evolution of Saprophagy-Related Genes in Brush-Footed Butterflies" International Journal of Molecular Sciences 24, no. 3: 2087. https://doi.org/10.3390/ijms24032087