

Targeting Cathepsin L in Cancer Management: Leveraging Machine Learning, Structure-Based Virtual Screening, and Molecular Dynamics Studies
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
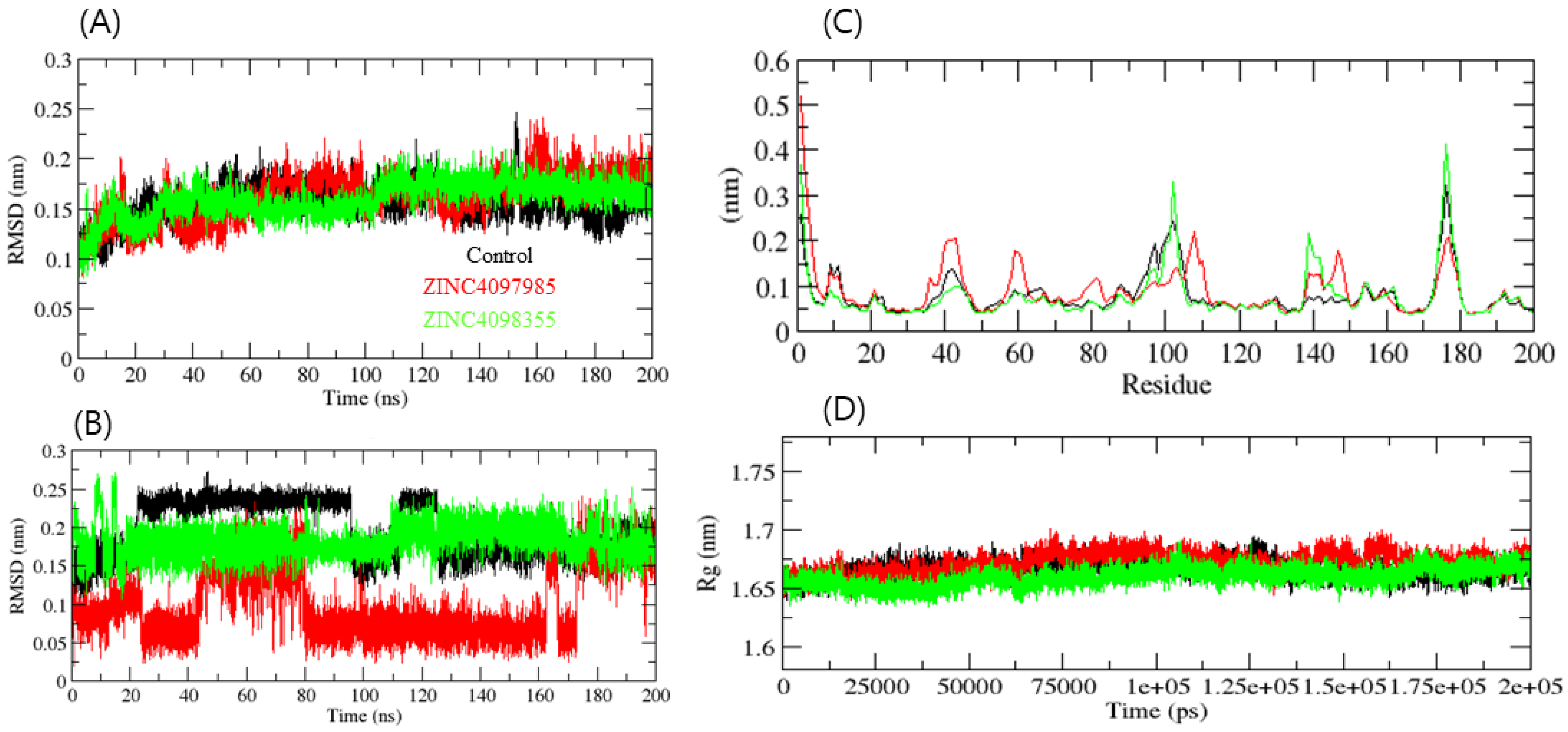
2. Results and Discussion
3. Methods
3.1. Machine Learning Model Building and Screening Natural Compounds
3.2. CTSL Protein Retrieval and Preparation
3.3. Structure-Based Virtual Screening
3.4. Physiochemical and ADMET Properties Prediction
3.5. Molecular Dynamics (MD) Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Fonovic, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta 2014, 1840, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Kukor, Z.; Mayerle, J.; Kruger, B.; Toth, M.; Steed, P.M.; Halangk, W.; Lerch, M.M.; Sahin-Toth, M. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J. Biol. Chem. 2002, 277, 21389–21396. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902. [Google Scholar] [CrossRef]
- Rudzinska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kondo, C.; Kojima, T.; Nagata, H.; Moriyama, A.; Hayakawa, T.; Katunuma, N. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother. Radiopharm. 2006, 21, 217–224. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed]
- Skrzydlewska, E.; Sulkowska, M.; Koda, M.; Sulkowski, S. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J. Gastroenterol. 2005, 11, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Sobotic, B.; Vizovisek, M.; Vidmar, R.; Van Damme, P.; Gocheva, V.; Joyce, J.A.; Gevaert, K.; Turk, V.; Turk, B.; Fonovic, M. Proteomic Identification of Cysteine Cathepsin Substrates Shed from the Surface of Cancer Cells. Mol. Cell Proteom. 2015, 14, 2213–2228. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes. Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.S.; Goldstein, L.J.; Gottesman, M.M. Expression of cathepsin L in human tumors. Cancer Res. 1991, 51, 1478–1481. [Google Scholar] [PubMed]
- Xu, X.; Yu, T.; Dong, L.; Glauben, R.; Wu, S.; Huang, R.; Qumu, S.; Chang, C.; Guo, J.; Pan, L.; et al. Eosinophils promote pulmonary matrix destruction and emphysema via Cathepsin L. Signal Transduct. Target. Ther. 2023, 8, 390. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Marquis, R.W.; James, I.; Zeng, J.; Trout, R.E.; Thompson, S.; Rahman, A.; Yamashita, D.S.; Xie, R.; Ru, Y.; Gress, C.J.; et al. Azepanone-based inhibitors of human cathepsin L. J. Med. Chem. 2005, 48, 6870–6878. [Google Scholar] [CrossRef]
- Kuhn, B.; Tichy, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef]
- Parker, E.N.; Song, J.; Kishore Kumar, G.D.; Odutola, S.O.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Strecker, T.E.; Barnes, A.L.; Sudhan, D.R.; Wittenborn, T.R.; et al. Synthesis and biochemical evaluation of benzoylbenzophenone thiosemicarbazone analogues as potent and selective inhibitors of cathepsin L. Bioorg. Med. Chem. 2015, 23, 6974–6992. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ketkar, R.; Tao, P. ADMETboost: A web server for accurate ADMET prediction. J. Mol. Model. 2022, 28, 408. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; The International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Zagidullin, B.; Wang, Z.; Guan, Y.; Pitkanen, E.; Tang, J. Comparative analysis of molecular fingerprints in prediction of drug combination effects. Brief. Bioinform. 2021, 22, bbab291. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 2014, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, H.; Yu, K.; Jin, Z. Molecular docking-based computational platform for high-throughput virtual screening. CCF Trans. High. Perform. Comput. 2022, 4, 63–74. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Showalter, S.A.; Bruschweiler, R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the AMBER99SB Force Field. J. Chem. Theory Comput. 2007, 3, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE–AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Yagasaki, T.; Matsumoto, M.; Tanaka, H. Lennard-Jones Parameters Determined to Reproduce the Solubility of NaCl and KCl in SPC/E, TIP3P, and TIP4P/2005 Water. J. Chem. Theory Comput. 2020, 16, 2460–2473. [Google Scholar] [CrossRef] [PubMed]
- Mencel, K.; Starynowicz, P.; Siczek, M.; Piecha-Bisiorek, A.; Jakubas, R.; Medycki, W. Symmetry breaking structural phase transitions, dielectric properties and molecular motions of formamidinium cations in 1D and 2D hybrid compounds: (NH2CHNH2)3[Bi2Cl9] and (NH2CHNH2)3[Bi2Br9]. Dalton Trans. 2019, 48, 14829–14838. [Google Scholar] [CrossRef]
- Mor, A.; Ziv, G.; Levy, Y. Simulations of proteins with inhomogeneous degrees of freedom: The effect of thermostats. J. Comput. Chem. 2008, 29, 1992–1998. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Binding Affinity (kcal/mol) | RF_Prediction Score | MolWt | MolLogP | HA | HD | RB |
|---|---|---|---|---|---|---|---|
| ZINC4097985 | −7.9 | 0.794152879 | 408.403 | −0.3084 | 9 | 4 | 4 |
| ZINC4098355 | −7.6 | 0.788373218 | 422.474 | 2.6621 | 8 | 0 | 8 |
| ZINC96023730 | −6.4 | 0.684678299 | 484.721 | 6.6687 | 4 | 1 | 10 |
| ZINC38429839 | −7.2 | 0.652458095 | 456.711 | 6.6289 | 3 | 2 | 5 |
| ZINC13383393 | −6.5 | 0.651968775 | 358.302 | 2.2031 | 6 | 5 | 4 |
| ZINC299817526 | −6.6 | 0.648805457 | 468.678 | 7.4357 | 2 | 2 | 9 |
| ZINC4098425 | −7.5 | 0.646199152 | 232.235 | 0.969 | 4 | 1 | 1 |
| ZINC5665355 | −6.6 | 0.643019115 | 470.694 | 7.6597 | 2 | 2 | 9 |
| ZINC1702729 | −6.9 | 0.640874436 | 388.416 | 2.8323 | 7 | 3 | 6 |
| ZINC238760072 | −6.5 | 0.633125885 | 376.493 | 4.5247 | 5 | 0 | 8 |
| ZINC238790964 | −6.4 | 0.624866697 | 430.541 | 2.3323 | 6 | 3 | 3 |
| ZINC28876559 | −6.4 | 0.621405139 | 440.712 | 7.2275 | 2 | 2 | 5 |
| ZINC3982483 | −6.6 | 0.620906903 | 406.545 | −0.857 | 8 | 5 | 7 |
| AZ12878478 (control) | −6.3 | - | - | - | - | - | - |
| Molecule Property | Value | Unit | ||
|---|---|---|---|---|
| ZINC4097985 | ZINC4098355 | |||
| Absorption | ||||
| Caco-2 Permeability | −5.51 | −5.27 | log(cm/s) | |
| HIA | 62.85 | 66.63 | % | |
| Pgp Inhibition | 36.88 | 30.23 | % | |
| log D7.4 | 1.83 | 1.72 | log-ratio | |
| Aqueous Solubility | −4.46 | −4.09 | log(mol/L) | |
| Oral Bioavailability | 36.66 | 40.58 | % | |
| Distribution | ||||
| BBB | 18.77 | 29.17 | % | |
| PPBR | 35.01 | 45.2 | % | |
| VDss | 3.13 | 3.39 | L/kg | |
| Metabolism | ||||
| CYP2C9 | Inhibition | 50.93 | 53.89 | % |
| CYP2D6 | 98.02 | 92.92 | ||
| CYP3A4 | 34.92 | 35.24 | ||
| CYP2C9 | Substrate | 34.14 | 32.66 | |
| CYP2D6 | 51.03 | 56.54 | ||
| CYP3A4 | 41.78 | 42.75 | ||
| Excretion | ||||
| Half-Life | 58.17 | 63.46 | h | |
| CL-Hepa | 47.36 | 55.3 | μL min−1 (106 cells)−1 | |
| CL-Micro | 40.61 | 48.28 | mL min−1 g−1 | |
| Toxicity | ||||
| hERG Blockers | 43.62 | 40.04 | % | |
| Ames | 48.32 | 39.31 | ||
| DILI | 49.79 | 59.98 | ||
| LD50 | 2.75 | 2.45 | −log(mol/kg) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almalki, A.A.; Shafie, A.; Hazazi, A.; Banjer, H.J.; Bakhuraysah, M.M.; Almaghrabi, S.A.; Alsaiari, A.A.; Alsaeedi, F.A.; Ashour, A.A.; Alharthi, A.; et al. Targeting Cathepsin L in Cancer Management: Leveraging Machine Learning, Structure-Based Virtual Screening, and Molecular Dynamics Studies. Int. J. Mol. Sci. 2023, 24, 17208. https://doi.org/10.3390/ijms242417208
Almalki AA, Shafie A, Hazazi A, Banjer HJ, Bakhuraysah MM, Almaghrabi SA, Alsaiari AA, Alsaeedi FA, Ashour AA, Alharthi A, et al. Targeting Cathepsin L in Cancer Management: Leveraging Machine Learning, Structure-Based Virtual Screening, and Molecular Dynamics Studies. International Journal of Molecular Sciences. 2023; 24(24):17208. https://doi.org/10.3390/ijms242417208
Chicago/Turabian StyleAlmalki, Abdulraheem Ali, Alaa Shafie, Ali Hazazi, Hamsa Jameel Banjer, Maha M. Bakhuraysah, Sarah Abdullah Almaghrabi, Ahad Amer Alsaiari, Fouzeyyah Ali Alsaeedi, Amal Adnan Ashour, Afaf Alharthi, and et al. 2023. "Targeting Cathepsin L in Cancer Management: Leveraging Machine Learning, Structure-Based Virtual Screening, and Molecular Dynamics Studies" International Journal of Molecular Sciences 24, no. 24: 17208. https://doi.org/10.3390/ijms242417208