

MetaSpot: A General Approach for Recognizing the Reactive Atoms Undergoing Metabolic Reactions Based on the MetaQSAR Database
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MetaQSAR-Based Datasets
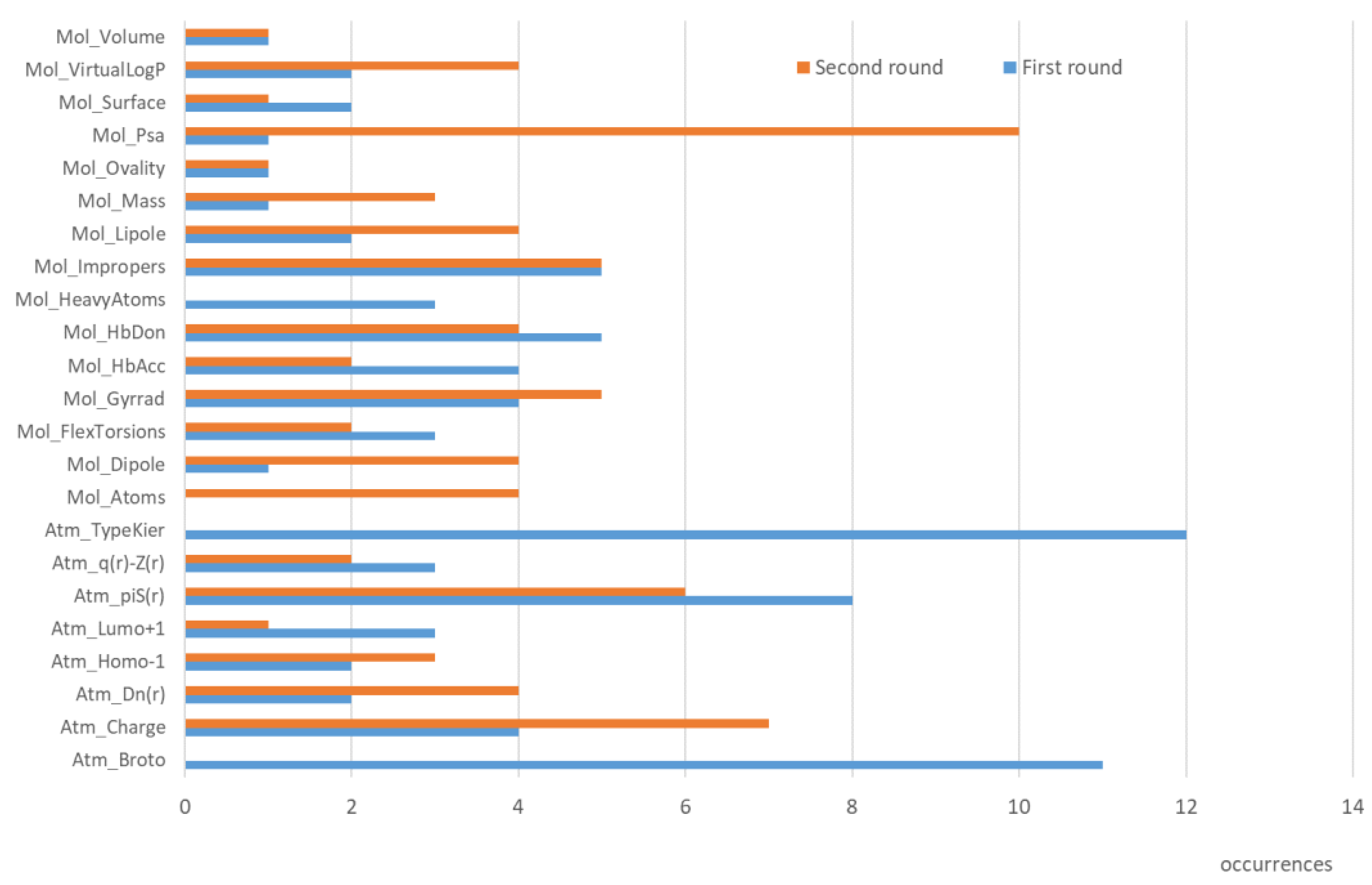
2.2. Prediction of the Reactive Atoms for the Reaction Classes
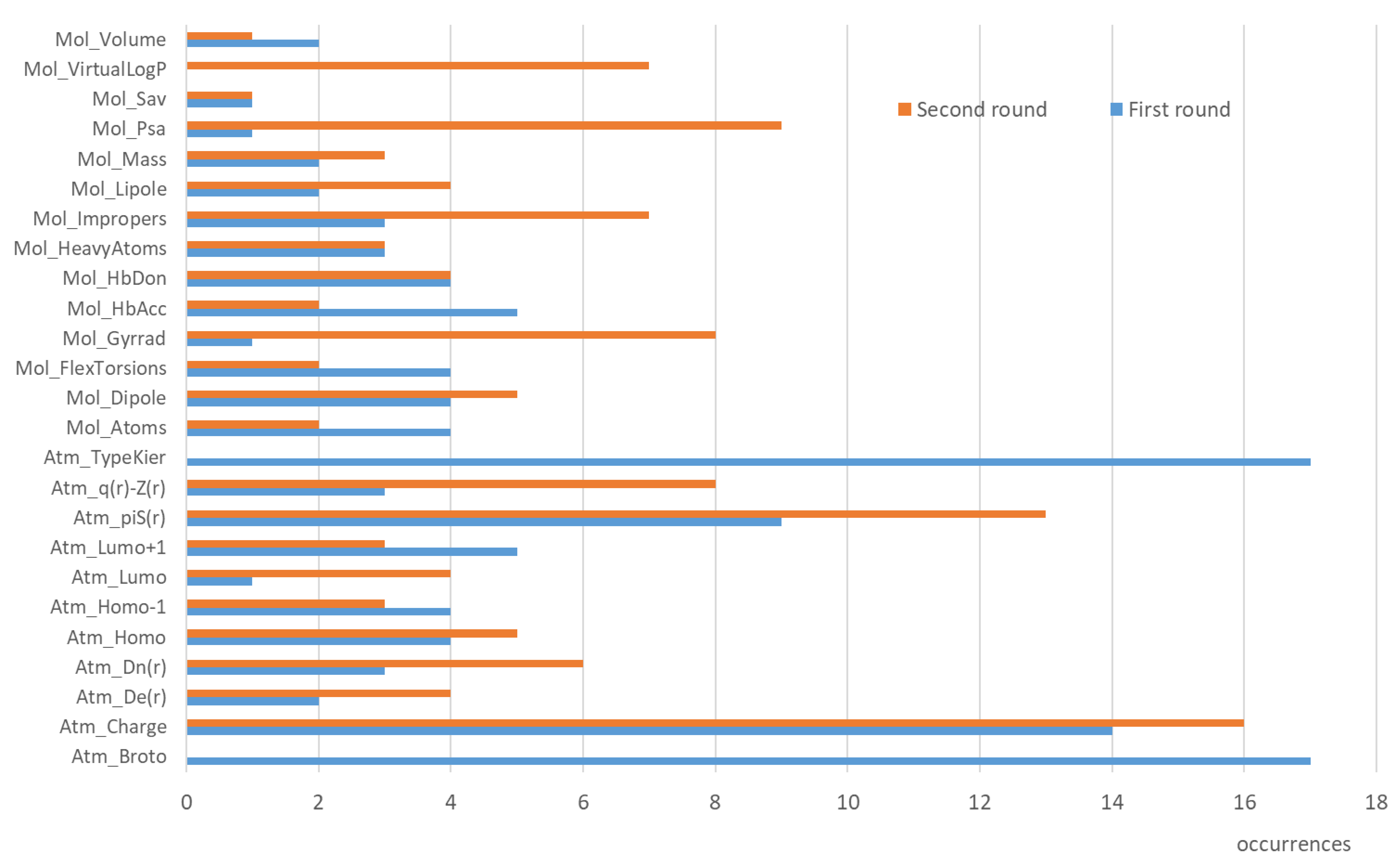
2.3. Prediction of the Reactive Atoms for the Reaction Subclasses
3. Discussion
- (1)
- Redox reactions on carbon atoms are conveniently predicted even without atom typing; this outcome indicates that their reactive atoms mostly depend on the considered stereo-electronic descriptors (e.g., atomic charges and self-polarizability).
- (2)
- Redox reactions involving nitrogen atoms (and to minor extent Sulphur atoms) are satisfactorily predicted only when using atom types, thus suggesting that their reactivity depends on different factors compared to carbon atoms.
- (3)
- Hydrolysis reactions yield markedly more accurate predictions when including atom types which reasonably allow an easy detection of the labile groups.
- (4)
- Conjugations with glucuronic acid are substantially unpredictable without atom types; this finding suggests that the reactivity of the involved centers depends on stereo-electronic factors not included in the considered descriptors.
- (5)
- Most reactions with glutathione can be successfully predicted even without atom types, a result which can be explained by considering that these conjugations depend on the here parameterized electrophilicity of the reactive centers.
4. Materials and Methods
4.1. Utilized Metabolic Data
4.2. Generation of Models with Atom Typing (First Round)
4.3. Generation of Models without Atom Typing (Second Round)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazmi, S.R.; Jun, R.; Yu, M.S.; Jung, C.; Na, D. In silico approaches and tools for the prediction of drug metabolism and fate: A review. Comput. Biol. Med. 2019, 106, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, S. In silico ADME-Tox modeling: Progress and prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, R.; Bharatam, P.V. Molecular Modeling Studies on Cytochrome P450-mediated Drug Metabolism. Curr. Drug Metab. 2021, 22, 683–697. [Google Scholar] [CrossRef]
- Ford, K.A.; Ryslik, G.; Sodhi, J.; Halladay, J.; Diaz, D.; Dambach, D.; Masuda, M. Computational predictions of the site of metabolism of cytochrome P450 2D6 substrates: Comparative analysis; molecular docking; bioactivation and toxicological implications. Drug Metab. Rev. 2015, 47, 291–319. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef]
- Long, A. Drug metabolism in silico—The knowledge-based expert system approach. Historical perspectives and current strategies. Drug Discov. Today Technol. 2013, 10, e147–e153. [Google Scholar] [CrossRef]
- Erhardt, P.W. A human drug metabolism database: Potential roles in the quantitative predictions of drug metabolism and metabolism-related drug-drug interactions. Curr. Drug Metab. 2003, 4, 411–422. [Google Scholar] [CrossRef]
- Litsa, E.E.; Das, P.; Kavraki, L.E. Machine learning models in the prediction of drug metabolism: Challenges and future perspectives. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tian, S.; Allen, D.; Oler, E.; Peters, H.; Lui, V.W.; Gautam, V.; Djoumbou-Feunang, Y.; Greiner, R.; Metz, T.O. BioTransformer 3.0-a web server for accurately predicting metabolic transformation products. Nucleic Acids Res. 2022, 50, W115–W123. [Google Scholar] [CrossRef] [PubMed]
- Šícho, M.; Stork, C.; Mazzolari, A.; de Bruyn Kops, C.; Pedretti, A.; Testa, B.; Vistoli, G.; Svozil, D.; Kirchmair, J. FAME 3: Predicting the Sites of Metabolism in Synthetic Compounds and Natural Products for Phase 1 and Phase 2 Metabolic Enzymes. J. Chem. Inf. Model. 2019, 59, 3400–3412. [Google Scholar] [CrossRef] [PubMed]
- Matlock, M.K.; Hughes, T.B.; Swamidass, S.J. XenoSite server: A web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. [Google Scholar] [CrossRef] [Green Version]
- Karp, P.D. Can we replace curation with information extraction software? Database 2016, 2016, baw150. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Vistoli, G.; Testa, B. MetaQSAR: An Integrated Database Engine to Manage and Analyze Metabolic Data. J. Med. Chem. 2018, 61, 1019–1030. [Google Scholar] [CrossRef]
- Mazzolari, A.; Sommaruga, L.; Pedretti, A.; Vistoli, G. MetaTREE, a Novel Database Focused on Metabolic Trees, Predicts an Important Detoxification Mechanism: The Glutathione Conjugation. Molecules 2021, 26, 2098. [Google Scholar] [CrossRef]
- Mazzolari, A.; Afzal, A.M.; Pedretti, A.; Testa, B.; Vistoli, G.; Bender, A. Prediction of UGT-mediated Metabolism Using the Manually Curated MetaQSAR Database. ACS Med. Chem. Lett. 2019, 10, 633–638. [Google Scholar] [CrossRef]
- Mazzolari, A.; Scaccabarozzi, A.; Vistoli, G.; Pedretti, A. MetaClass, a Comprehensive Classification System for Predicting the Occurrence of Metabolic Reactions Based on the MetaQSAR Database. Molecules 2021, 26, 5857. [Google Scholar] [CrossRef]
- Hall, L.H.; Mohney, B.; Kier, L.B. The Electrotopological State: Structure Information at the Atomic Level for Molecular Graphs. J. Chem. Inf. Comput. Sci. 1991, 31, 76–82. [Google Scholar] [CrossRef]
- Broto, P.; Moreau, G.; Vandycke, C. Molecular structures: Perception, autocorrelation descriptor and sar studies. Eur. J. Med. Chem. 1984, 19, 66–70. [Google Scholar]
- Hughes, T.B.; Miller, G.P.; Swamidass, S.J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione. Chem. Res. Toxicol. 2015, 28, 797–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: A versatile platform for cheminformatics and drug design projects. Bioinformatics 2020, 37, 1174–1175. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software. ACM SIGKDD Explor. Newsletter 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Tree2C: A Flexible Tool for Enabling Model Deployment with Special Focus on Cheminformatics Applications. Appl. Sci. 2020, 10, 7704. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzolari, A.; Perazzoni, P.; Sabato, E.; Lunghini, F.; Beccari, A.R.; Vistoli, G.; Pedretti, A. MetaSpot: A General Approach for Recognizing the Reactive Atoms Undergoing Metabolic Reactions Based on the MetaQSAR Database. Int. J. Mol. Sci. 2023, 24, 11064. https://doi.org/10.3390/ijms241311064
Mazzolari A, Perazzoni P, Sabato E, Lunghini F, Beccari AR, Vistoli G, Pedretti A. MetaSpot: A General Approach for Recognizing the Reactive Atoms Undergoing Metabolic Reactions Based on the MetaQSAR Database. International Journal of Molecular Sciences. 2023; 24(13):11064. https://doi.org/10.3390/ijms241311064
Chicago/Turabian StyleMazzolari, Angelica, Pietro Perazzoni, Emanuela Sabato, Filippo Lunghini, Andrea R. Beccari, Giulio Vistoli, and Alessandro Pedretti. 2023. "MetaSpot: A General Approach for Recognizing the Reactive Atoms Undergoing Metabolic Reactions Based on the MetaQSAR Database" International Journal of Molecular Sciences 24, no. 13: 11064. https://doi.org/10.3390/ijms241311064