
Integrative Analysis of Metabolome and Transcriptome Provides Insights into the Mechanism of Flower Induction in Pineapple (Ananas comosus (L.) Merr.) by Ethephon

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
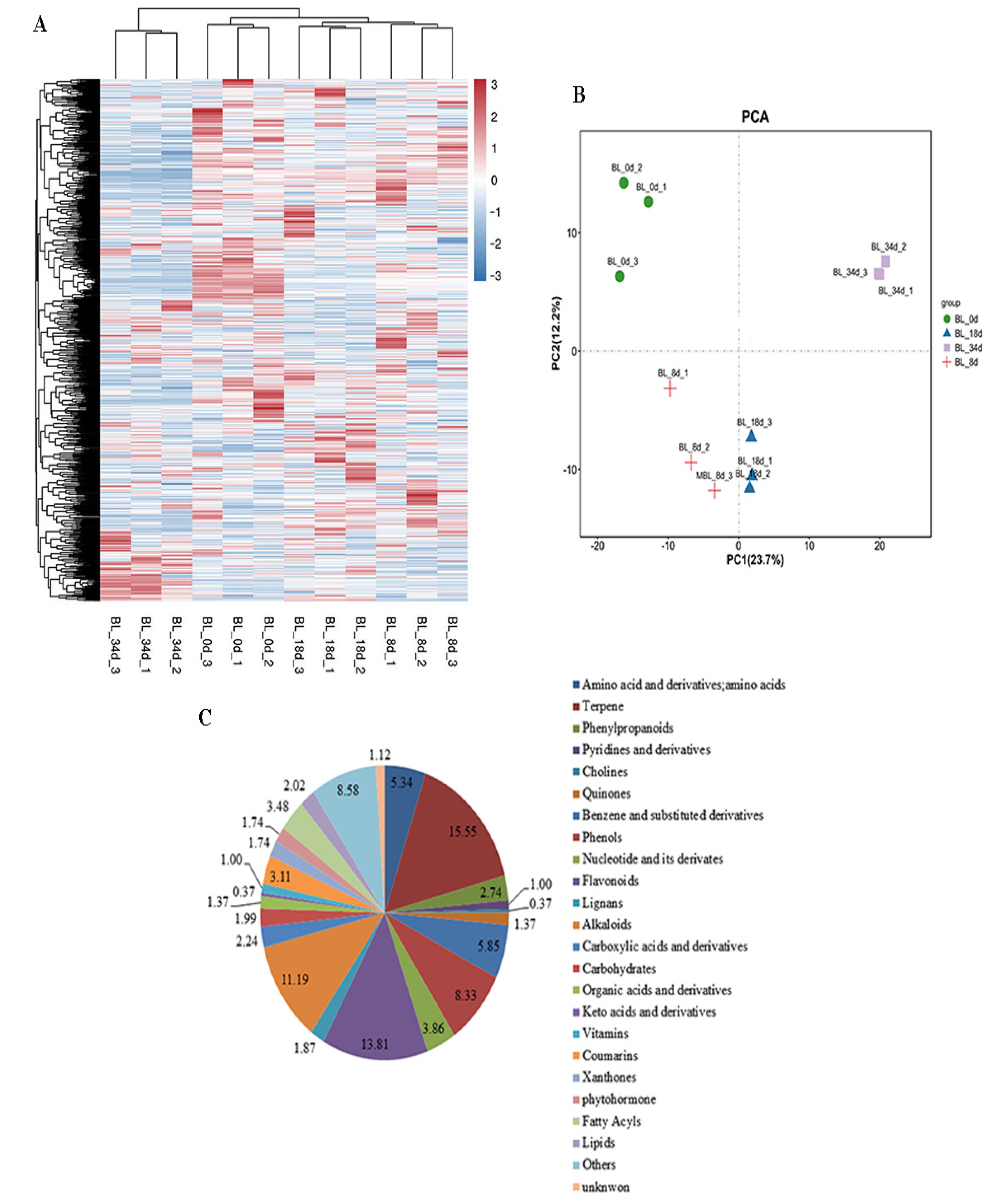
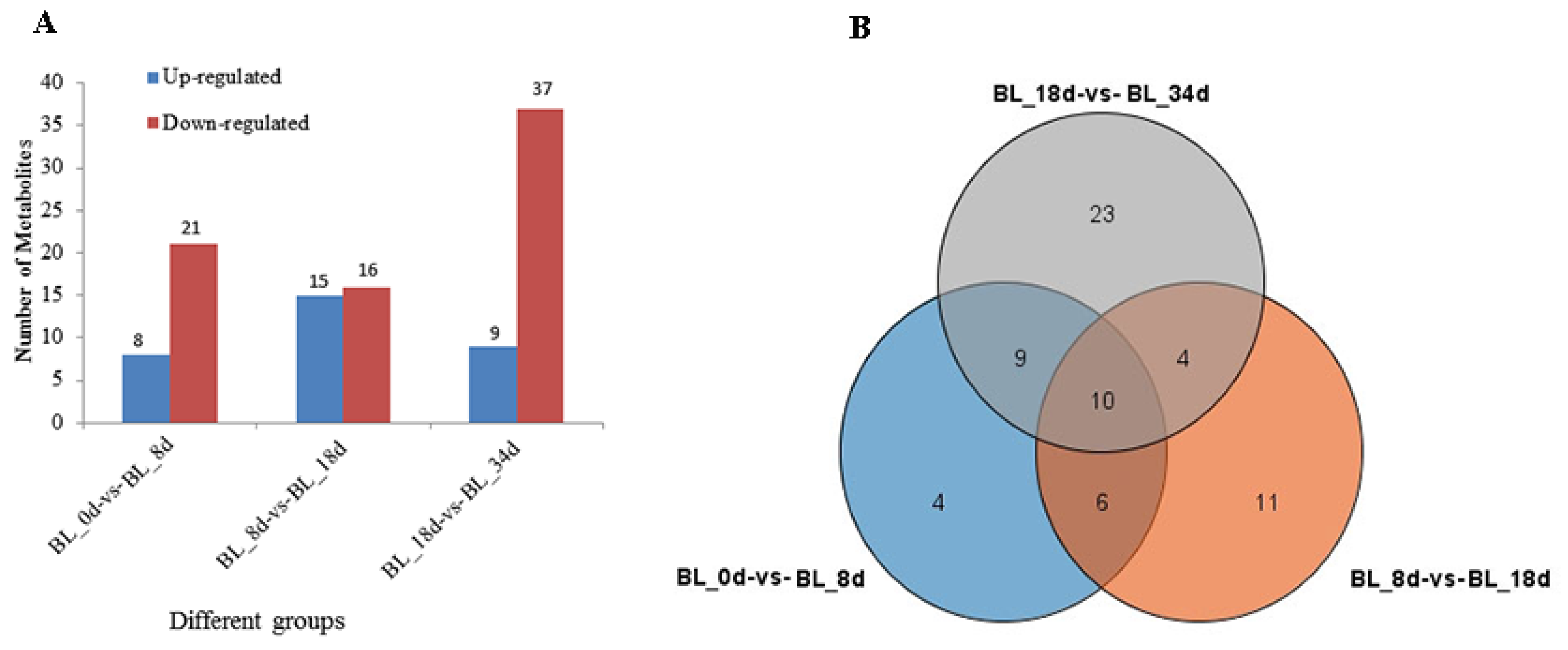
2.1. Overview of Metabolite Profiles during Flower Induction
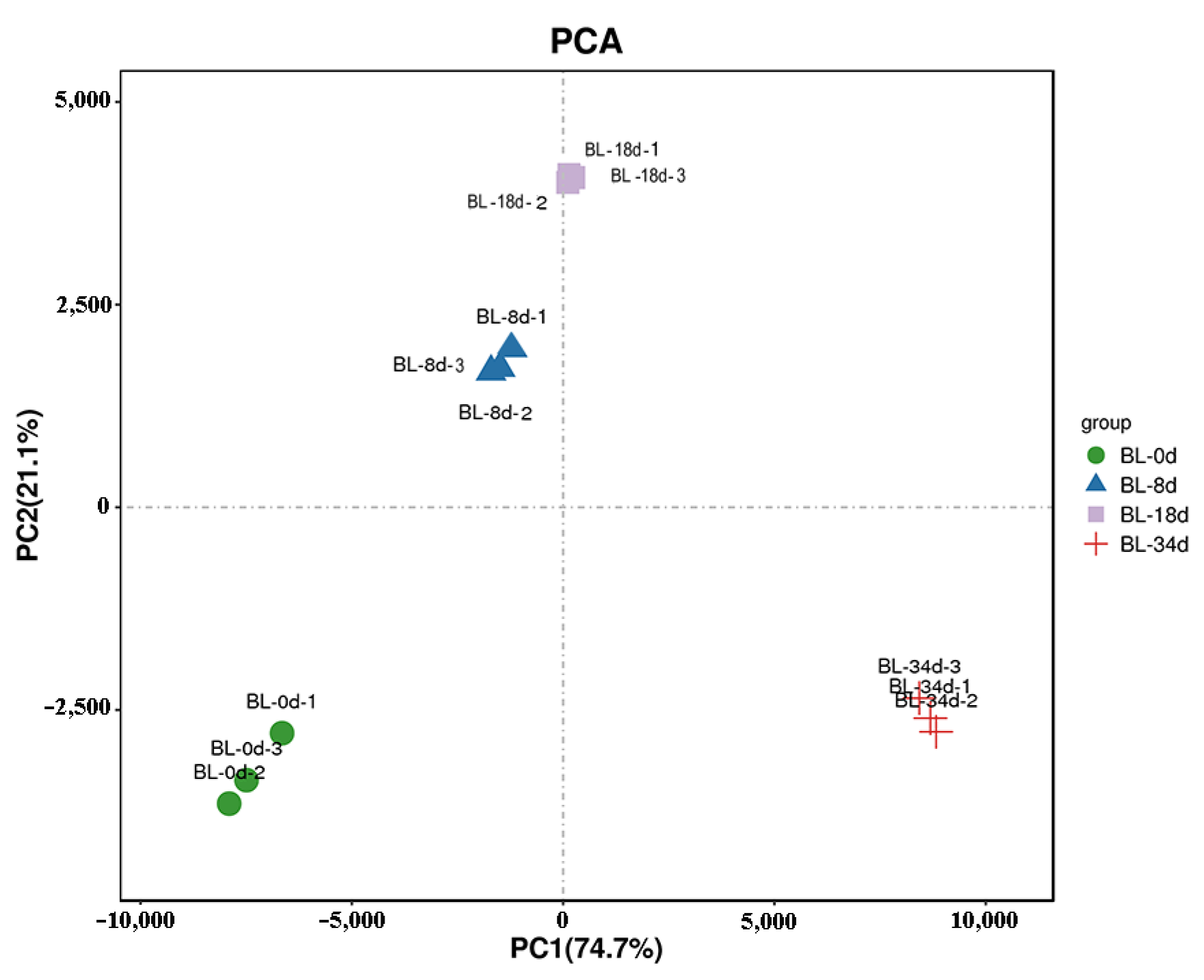
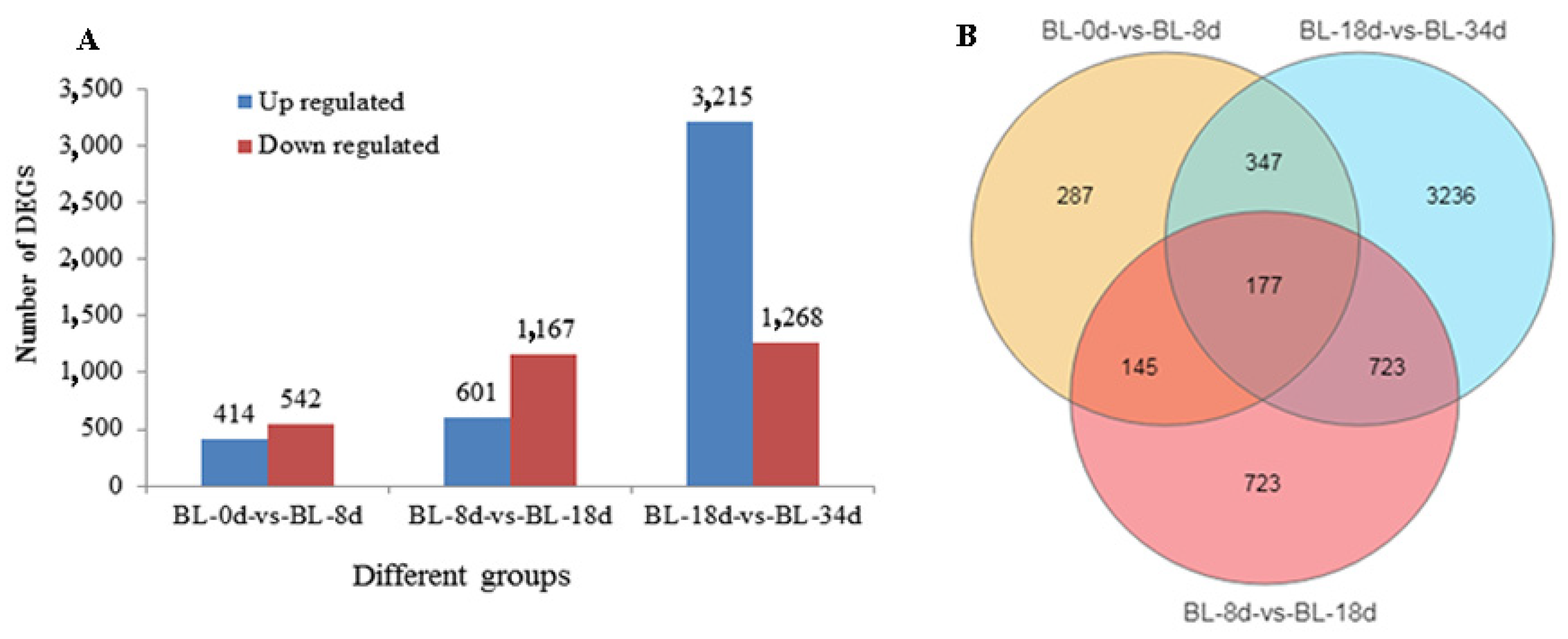
2.2. Transcriptome Analyses
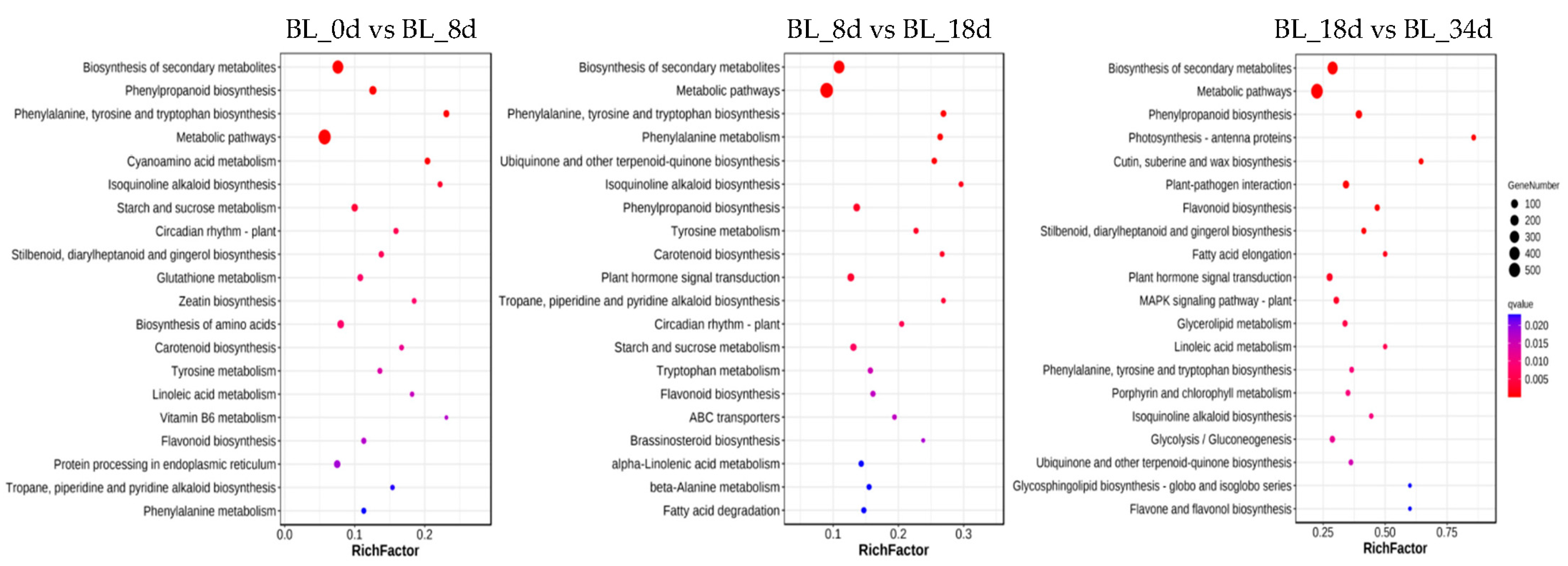
2.3. Functional Annotation of Differentially Expressed Genes
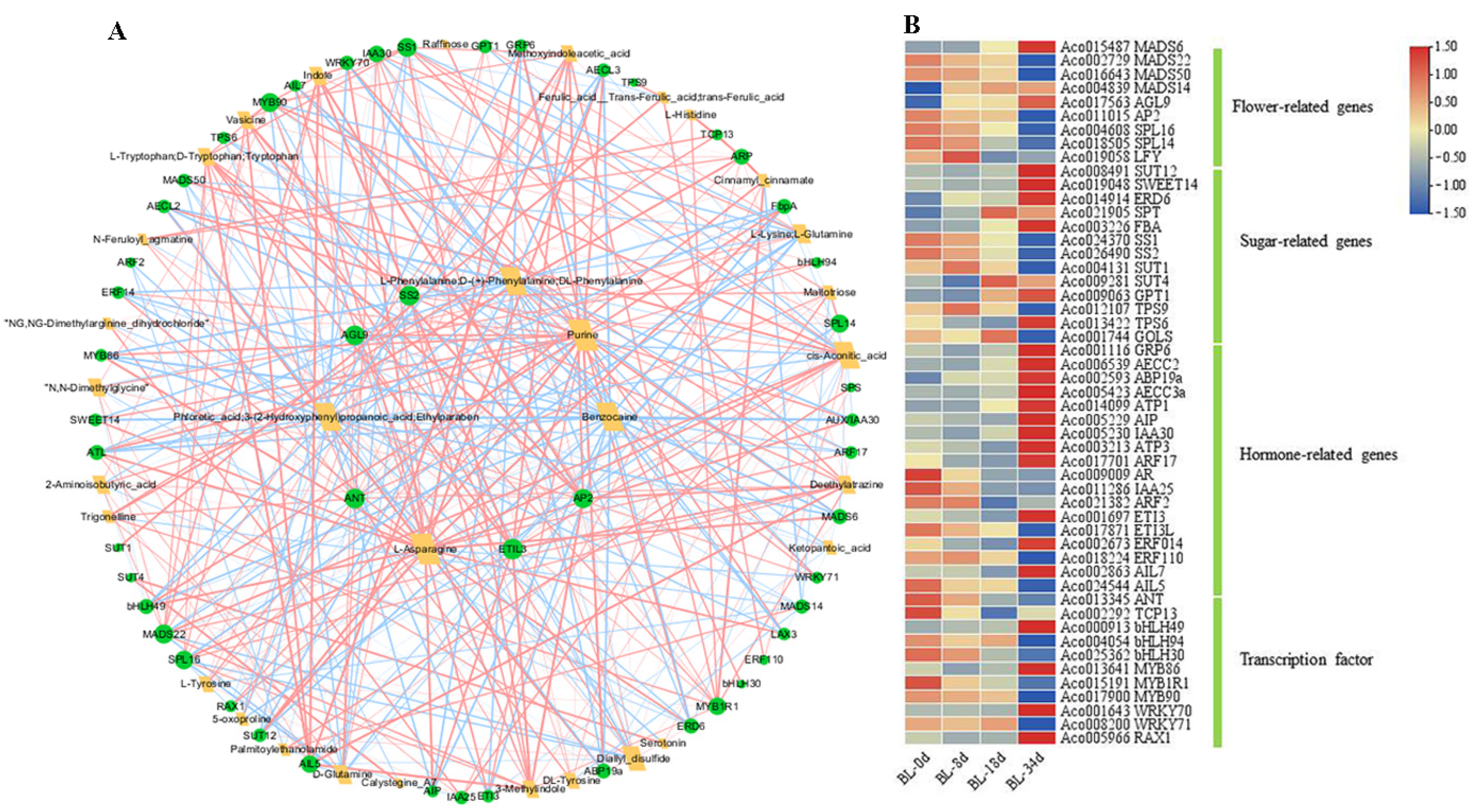
2.4. Integrative Analysis of DAMs and DEGs
2.5. Verification of DEGs by qPCR
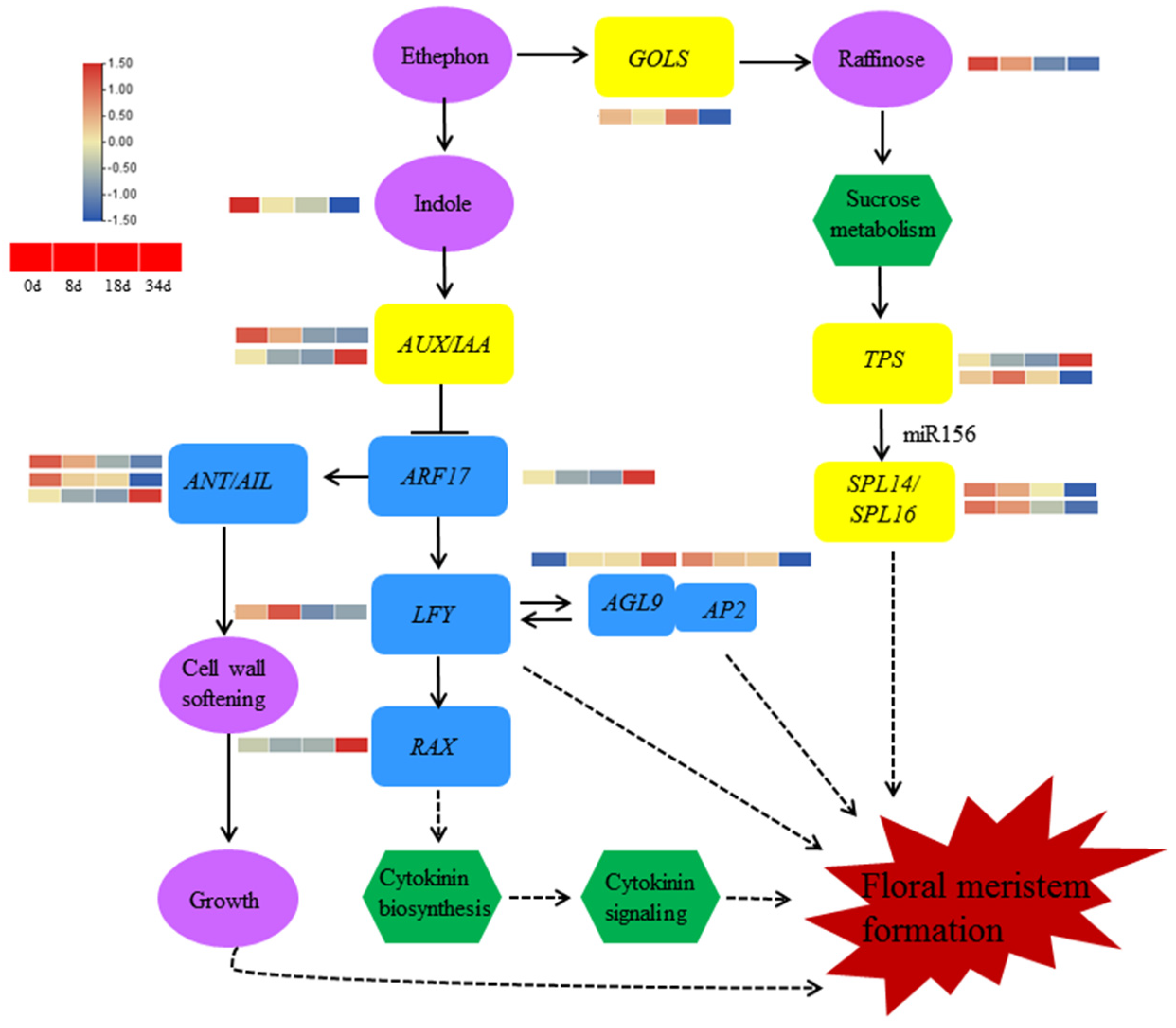
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Metabolomic Analysis
4.2.1. Sample Preparation and LC–ESI-MS/MS System Analysis
4.2.2. Differentially Accumulated Metabolites Analysis
4.3. RNA-seq Analysis
4.3.1. RNA Isolation and Library Construction
4.3.2. Differentially Expressed Genes (DEGs) Analysis
4.4. Association Analysis between Transcriptome and Metabolome
4.5. qRT-PCR Verification of Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BL | ‘Comte de Paris’ |
| qRT-PCR | Real-time quantitative reverse transcription polymerase chain reaction |
References
- Kurokura, T.; Mimida, N.; Battey, N.H.; Hytönen, T. The regulation of seasonal flowering in the Rosaceae. J. Exp. Bot. 2013, 64, 4131–4141. [Google Scholar] [CrossRef]
- Lin, Y.L.; Tsay, Y.F. Influence of differing nitrate and nitrogen availability on flowering control in Arabidopsis. J. Exp. Bot. 2017, 68, 2603–2609. [Google Scholar] [CrossRef]
- Liu, L.; Xuan, L.; Jiang, Y. Regulation by FLOWERING LOCUS T and TERMINAL FLOWER 1 in flowering time and plant architecture. Small Struct. 2021, 2, 2000125. [Google Scholar] [CrossRef]
- Siriwardana, N.S.; Lamb, R.S. The poetry of reproduction: The role of LEAFY in Arabidopsis thaliana flower formation. Int. J. Dev. Biol. 2012, 56, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.W.; Werr, W. A phylogenetically conserved APETALA2/ETHYLENE RESPONSE FACTOR, ERF12, regulates Arabidopsis floral development. Plant Mol. Biol. 2020, 102, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.C.; Hileman, L.C. SQUAMOSA-PROMOTER BINDING PROTEIN 1 initiates flowering in Antirrhinum majus through the activation of meristem identity genes. Plant J. 2010, 62, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A. Regulation of flowering by Trehalose-6-Phosphate signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef]
- Andrés, F.; Kinoshita, A.; Kalluri, N.; Fernández, V.; Falavigna, V.S.; Cruz, T.M.D.; Jang, S.; Chiba, Y.; Seo, M.; Mettler-Altmann, T.; et al. The sugar transporter SWEET10 acts downstream of FLOWERING LOCUS T during floral transition of Arabidopsis thaliana. BMC Plant Biol. 2020, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.H.; Pasriga, R.; Yoon, J.; Jeon, J.S.; An, G. Roles of sugars in controlling flowering time. J. Plant Biol. 2018, 61, 121–130. [Google Scholar] [CrossRef]
- Lu, B.; Chen, L.N.; Hao, J.B.; Zhang, Y.; Huang, J.C. Comparative transcription profiles reveal that carbohydrates and hormone signaling pathways mediate flower induction in Juglans sigillata after girdling. Ind. Crops Prod. 2020, 153, 112556. [Google Scholar] [CrossRef]
- Chakraborty, A.; Chaudhury, R.; Dutta, S.; Basak, M.; Dey, S.; Schäffner, A.R.; Das, M. Role of metabolites in flower development and discovery of compounds controlling flowering time. Plant Physiol. Bioch. 2022, 190, 109–118. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Sarnowska, E.; Nagy, F.; Davis, S.J. Genetic analyses of interactions among gibberellin, abscisic acid, and brassinosteroids in the control of flowering time in Arabidopsis thaliana. PLoS ONE 2010, 5, e14012. [Google Scholar] [CrossRef]
- Matsoukas, I.G. Interplay between sugar and hormone signaling pathways modulate floral signal transduction. Front. Genet. 2014, 5, 218. [Google Scholar] [CrossRef] [PubMed]
- Mutasa-Göttgens, E.; Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 2009, 60, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Amasino, R. Seasonal and developmental timing of flowering. Plant J. 2010, 61, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Montaigu, A.; Tóth, R.; Coupland, G. Plant development goes like clockwork. Trends Genet. 2010, 26, 296–306. [Google Scholar] [CrossRef]
- Liu, S.H.; Zang, X.P.; Sun, G.M. Changes in endogenous hormone concentrations during inflorescence induction and development in pineapple (Ananas comosus cv. Smooth Cayenne) by ethephon. Acad. J. 2011, 11, 124. [Google Scholar]
- Silva, G.F.; Silva, E.M.; Correa, J.P.; Vicente, M.H.; Jiang, N.; Notini, M.M.; Junior, A.C.; De Jesus, F.A.; Castilho, P.; Carrera, E.; et al. Tomato floral induction and flower development are orchestrated by the interplay between gibberellin and two unrelated microRNA-controlled modules. New Phytol. 2019, 221, 1328–1344. [Google Scholar] [CrossRef]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef]
- Dong, X.X.; Li, Y.J.; Guan, Y.H.; Wang, S.X.; Luo, H.; Li, X.M.; Li, H.; Zhang, Z.H. Auxin-induced AUXIN RESPONSE FACTOR4 activates APETALA1 and FRUITFULL to promote flowering in woodland strawberry. Hortic. Res. 2021, 8, 115. [Google Scholar] [CrossRef]
- Gazzarrini, S.; Tsai, A.Y.L. Trehalose-6-phosphate and SnRK1 kinases in plant development and signaling: The emerging picture. Plant Physiol. 2014, 5, 119. [Google Scholar]
- Shu, K.; Luo, X.; Meng, Y.; Yang, W. Toward a molecular understanding of abscisic acid actions in floral transition. Plant Cell Physiol. 2018, 59, 215–221. [Google Scholar] [CrossRef]
- Kazan, K.; Lyons, R. The link between flowering time and stress tolerance. J. Exp. Bot. 2016, 67, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Takeno, K. Stress and salicylic acid induce the expression of PnFT2 in the regulation of the stress-induced flowering of Pharbitis nil. J. Plant Physiol. 2014, 171, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, M.E.A.; Moreira, R.O.; Lima, A.A.; Ságio, S.A.; Barreto, H.G.; Luiz, S.L.P.; Abreu, C.E.A.; Yanes-Paz, E.; Ruíz, Y.C.; González-Olmedo, J.L.; et al. Early histological, hormonal, and molecular changes during pineapple (Ananas comosus (L.) Merrill) artificial flowering induction. J. Plant Physiol. 2017, 209, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, Q.S.; Liu, S.H.; Zhang, H.N.; Lin, W.Q.; Zhang, X.M.; Li, Y.H. Combining single molecule sequencing and Illumina RNA sequencing to elucidate flower induction of pineapple (Ananas comosus (L.) Merr.) treated with exogenous ethylene. Plant Growth Regul. 2021, 94, 303–321. [Google Scholar] [CrossRef]
- Liu, C.H.; Fan, C. De novo transcriptome assembly of floral buds of pineapple and identification of differentially expressed genes in response to ethephon induction. Front. Plant Sci. 2016, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Trusov, Y.; Botella, R.J. Silencing of the ACC synthase gene ACACS2 causes delayed flowering in pineapple [Ananas comosus (L.) Merr.]. J. Exp. Bot. 2006, 57, 3953–3960. [Google Scholar] [CrossRef]
- Ruan, C.C.; Chen, Z.; Hu, F.C.; Fan, W.; Wang, X.H.; Guo, L.J.; Fan, H.Y.; Luo, Z.W.; Zhang, Z.L. Genome-wide characterization and expression profiling of B3 superfamily during ethylene-induced flowering in pineapple (Ananas comosus L.). BMC Genom. 2021, 22, 561. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, X.; Liu, S.; Lin, W.; Li, Y.; Zhang, X. Genome-wide analysis of AP2/ERF transcription factors in pineapple reveals functional divergence during flowering induction mediated by ethylene and floral organ development. Genomics 2021, 113, 474–489. [Google Scholar] [CrossRef]
- Denay, G.; Chahtane, H.; Tichtinsky, G.; Parcy, F. A flower is born: An update on Arabidopsis floral meristem formation. Curr. Opin. Plant Biol. 2017, 35, 15–22. [Google Scholar] [CrossRef]
- Kumari, U.; Sengupta, K.K.S.; Misra, S.; Tiwary, A.K.; Lal, H.C.; Kumar, K. Studies on different growth regulators and stage of application on inflorescence induction and development of pineapple (Ananas comosus L. Merr.) Var. Queen. Int. J. Chem. Stud. 2020, 8, 1093–1095. [Google Scholar] [CrossRef]
- Conti, L. Hormonal control of the floral transition: Can one catch them all? Dev. Biol. 2017, 430, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.Y.; Li, J.; Guo, B.Y.; Li, L.; Ma, G.H.; Wu, K.L.; Yang, F.X.; Zhu, G.F.; Fang, L.; Zeng, S.J. Exogenous GA3 promotes flowering in paphiopedilum callosum (Orchidaceae) through bolting and lateral flower development regulation. Hortic. Res. 2022, 9, 91. [Google Scholar] [CrossRef]
- Singh, D.; Sharma, S.; Jose-Santhi, J.; Kalia, D.; Singh, R.K. Hormones regulate the flowering process in saffron differently depending on the developmental stage. Front. Plant Sci. 2023, 14, 1107172. [Google Scholar] [CrossRef]
- Cheng, H.; Zhou, M.; Si, Y.Y.; Li, W.J.; Wang, L.K.; Chen, S.M.; Chen, F.D.; Jiang, J.F. Transcriptome analysis of ethylene response in chrysanthemum moriflolium Ramat. with an emphasis on flowering delay. Horticulturae 2023, 9, 428. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.; Zhang, H.; Chen, L.; Yu, D. ERF1 delays flowering through direct inhibition of FLOWERING LOCUS T expression in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Xing, X.J.; Tang, Y.; Jin, J.Y.; Ding, L.; Song, A.P.; Chen, S.M.; Chen, F.D.; Jiang, J.F.; Fang, W.M. An ethylene-responsive transcription factor and a flowering locus KH domain homologue jointly modulate photoperiodic flowering in chrysanthemum. Plant Cell Environ. 2022, 45, 1442–1456. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Berns, M.C.; Nole-Wilson, S.; Yamaguchi, A.; Coupland, G.; Krizek, B.; Wagner, D. A molecular framework for auxin-mediated initiation of flower primordia. Dev. Cell 2013, 24, 271–282. [Google Scholar] [CrossRef]
- Wu, M.-F.; Yamaguchi, N.; Xiao, J.; Bargmann, B.; Estelle, M.; Sang, Y.; Doris, W. Auxin-regulated chromatin switch directs acquisition of flower primordium founder fate. eLife 2015, 4, e09269. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Sui, C.; Wang, S.H.; Liu, X.Q.; Zhang, G.X.; Sun, H.N.; Wan, K.; Yan, J.; Guo, S.J. Genome-wide evolutionary analysis of AUX/IAA gene family in wheat identifies a novel gene TaIAA15-1A regulating flowering time by interacting with ARF. Int. J. Biol. Macromol. 2023, 227, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Jose-Santhi, J.; Sheikh, F.R.; Kalia, D.; Singh, R.K. Sugar metabolism mediates temperature-dependent flowering induction in saffron (Crocus sativus L.). Environ. Exp. Bot. 2023, 206, 105150. [Google Scholar] [CrossRef]
- Gu, J.H.; Zeng, Z.; Wang, Y.R.; Lyu, Y.M. Transcriptome analysis of carbohydrate metabolism genes and molecular regulation of sucrose transport gene loSUT on the flowering process of developing oriental hybrid lily ‘sorbonne’ bulb. Int. J. Mol. Sci. 2020, 21, 3092. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, F.; Olas, J.J.; Feil, R.; Watanabe, M.; Krause, U.; Hoefgen, R.; Stitt, M.; Lunn, J.E. Functional features of TREHALOSE-6-PHOSPHATE SYNTHASE1-an essential enzyme in Arabidopsis thaliana. Plant Cell 2020, 32, 1949–1972. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.B.; Zhang, D.; Li, Y.M.; Shen, Y.W.; Zhao, C.P.; Ma, J.J.; An, N.; Han, M.Y. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in Apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Cui, Y.; Dong, R.; Luo, D.; Fang, L.; Nan, Z.; Liu, Z. Integrative analyses of transcriptomes and metabolomes reveal associated genes and metabolites with flowering regulation in common Vetch (Vicia sativa L.). Int. J. Mol. Sci. 2022, 23, 6818. [Google Scholar] [CrossRef]
- Kinoshita, A.; Richter, R. Genetic and molecular basis of floral induction in Arabidopsis thaliana. J. Exp. Bot. 2020, 7, 2490–2504. [Google Scholar] [CrossRef]
- Lin, W.Q.; Pu, Y.; Liu, S.H.; Wu, Q.S.; Yao, Y.L.; Yang, Y.M.; Zhang, X.M.; Sun, W.S. Genome-wide identification and expression patterns of AcSWEET family in pineapple and AcSWEET11 mediated sugar accumulation. Int. J. Mol. Sci. 2022, 23, 13875. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Liu, S.; Xiao, X.; Sun, W.; Lu, X.; Gao, Y.; He, J.; Zhu, Z.; Wu, Q.; Zhang, X. Integrative Analysis of Metabolome and Transcriptome Provides Insights into the Mechanism of Flower Induction in Pineapple (Ananas comosus (L.) Merr.) by Ethephon. Int. J. Mol. Sci. 2023, 24, 17133. https://doi.org/10.3390/ijms242417133
Lin W, Liu S, Xiao X, Sun W, Lu X, Gao Y, He J, Zhu Z, Wu Q, Zhang X. Integrative Analysis of Metabolome and Transcriptome Provides Insights into the Mechanism of Flower Induction in Pineapple (Ananas comosus (L.) Merr.) by Ethephon. International Journal of Molecular Sciences. 2023; 24(24):17133. https://doi.org/10.3390/ijms242417133
Chicago/Turabian StyleLin, Wenqiu, Shenghui Liu, Xiou Xiao, Weisheng Sun, Xinhua Lu, Yuyao Gao, Junjun He, Zhuying Zhu, Qingsong Wu, and Xiumei Zhang. 2023. "Integrative Analysis of Metabolome and Transcriptome Provides Insights into the Mechanism of Flower Induction in Pineapple (Ananas comosus (L.) Merr.) by Ethephon" International Journal of Molecular Sciences 24, no. 24: 17133. https://doi.org/10.3390/ijms242417133