
Micronutrient Deficiency in Inherited Metabolic Disorders Requiring Diet Regimen: A Brief Critical Review

 , and
, and
Abstract
:1. Introduction
2. Aim of the Study and Strategy Search
3. Micronutrients, Their Nutritional Sources, and Their Function
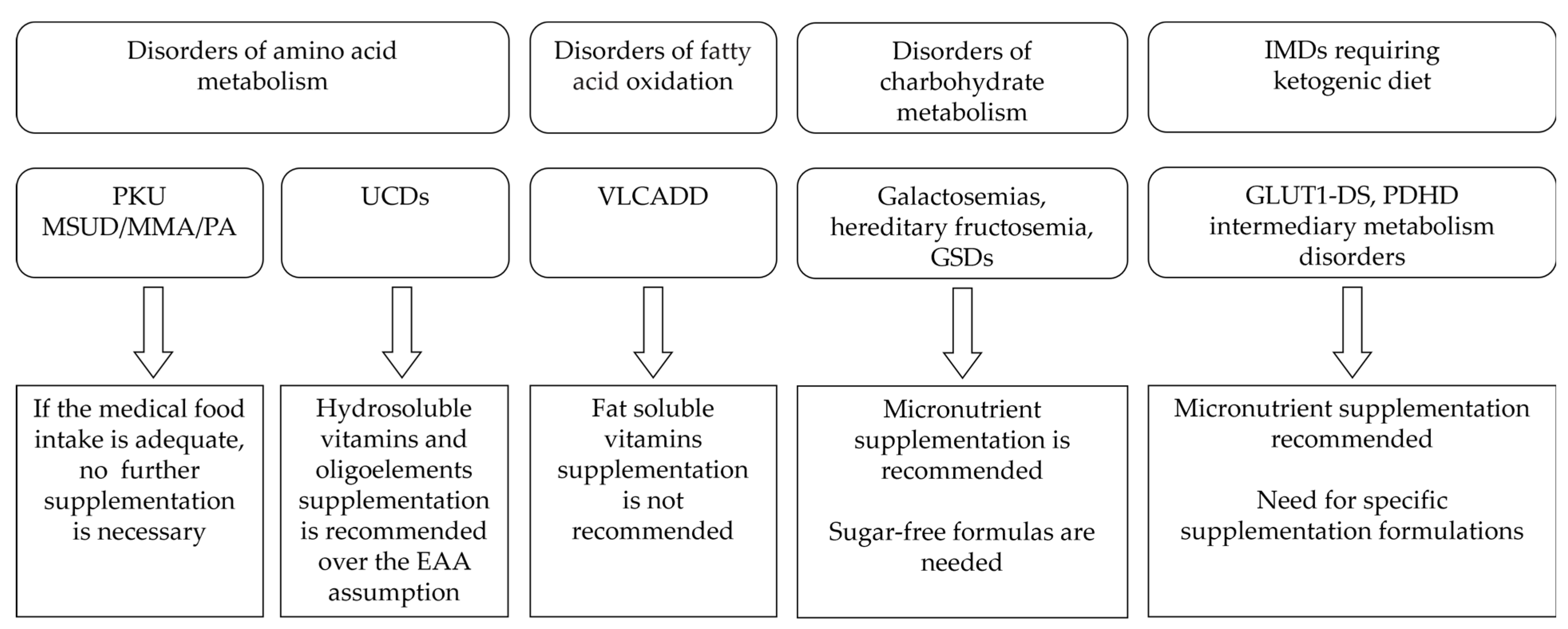
4. IMDs Requiring Special Diets
4.1. Disorders of Amino acid Metabolism
4.1.1. Phenylketonuria (PKU)
4.1.2. Maple Syrup Urine Disease, Propionic and Methylmalonic Acidemia
4.1.3. Urea Cycle Disorders
4.2. Disorders of Fatty Acid Oxidation
4.3. Disorders of Carbohydrate Metabolism
4.3.1. Galactosemias
4.3.2. Hereditary Fructosemia
4.3.3. Glycogen Storage Disorders (GSDs)
4.4. IMD Requiring Ketogenic Diet
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DRI | dietary reference intakes |
| EAA | essential amino acid |
| FAD | flavin adenine dinucleotide |
| FAOD | fatty acid oxidation disorder |
| FMN | flavin mononucleotide |
| GALT | galactose 1-phosphate urydyltransferase |
| GLUT1-DS | glucose transporter type 1 deficiency syndrome |
| GMP | glycomacropeptides |
| GSD | glycogen storage disorder |
| HF | hereditary fructosemia |
| IMD | inherited metabolic disorder |
| KD | ketogenic diet |
| LCFAOD | long-chain fatty acid oxidation disorder |
| MCT | medium-chain triglyceride |
| MCV | mean corpuscular volume |
| MMA | methylmalonic acidemia |
| MSUD | maple syrup urine disease |
| NE | niacin equivalents |
| OA | organic acidosis |
| PA | propionic acidemia |
| DHA | docosahexaenoic acid |
| PAH | phenylalanine hydroxylase |
| PDHc | pyruvate dehydrogenase complex |
| PKU | phenylketonuria |
| PLP | pyridoxal 5′-phosphate |
| RDA | recommended dietary allowances |
| UCD | urea cycle disorder |
| VLCADD | very-long-chain Acyl CoA dehydrogenase deficiency |
| WHO | World Health Organization |
References
- Berger, M.M.; Shenkin, A.; Schweinlin, A.; Amrein, K.; Augsburger, M.; Biesalski, H.K.; Bischoff, S.C.; Casaer, M.P.; Gundogan, K.; Lepp, H.L.; et al. ESPEN micronutrient guideline. Clin. Nutr. 2022, 41, 1357–1424. [Google Scholar] [CrossRef] [PubMed]
- Mehri, A. Trace Elements in Human Nutrition (II)—An Update. Int. J. Prev. Med. 2020, 11, 2. [Google Scholar] [PubMed]
- Shenkin, A. The key role of micronutrients. Clin. Nutr. 2006, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; de Benoist, B.; Dary, O.; Hurrel, R. (Eds.) Guidelines on Food Fortification with Micronutrients; World Health Organization (WHO): Geneva, Switzerland; Food and Agricultural Organization (FAO) of the United Nations: Rome, Italy, 2006.
- Battaglia-Hsu, S.F.; Ghemrawi, R.; Coelho, D.; Dreumont, N.; Mosca, P.; Hergalant, S.; Gauchotte, G.; Sequeira, J.M.; Ndiongue, M.; Houlgatte, R.; et al. Inherited disorders of cobalamin metabolism disrupt nucleocytoplasmic transport of mRNA through impaired methylation/phosphorylation of ELAVL1/HuR. Nucleic Acids Res. 2018, 46, 7844–7857. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Christodoulou, J.; Rahman, S. Disorders of riboflavin metabolism. J. Inherit. Metab. Dis. 2019, 42, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Leon-Del-Rio, A. Biotin in metabolism, gene expression, and human disease. J. Inherit. Metab. Dis. 2019, 42, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders affecting vitamin B(6) metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef]
- Tolomeo, M.; Nisco, A.; Leone, P.; Barile, M. Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. Int. J. Mol. Sci. 2020, 21, 5310. [Google Scholar] [CrossRef]
- MaCdonald, A.; van Rijn, M.; Feillet, F.; Lund, A.M.; Bernstein, L.; Bosch, A.M.; Gizewska, M.; van Spronsen, F.J. Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann. Nutr. Metab. 2012, 61, 289–295. [Google Scholar] [CrossRef]
- Singh, R.H. Nutritional management of patients with urea cycle disorders. J. Inherit. Metab. Dis. 2007, 30, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Belanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Gizewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Forny, P.; Horster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grunert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef] [PubMed]
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.L., 2nd; MacLeod, E.; Jurecka, A.; Hainline, B. Clinical manifestations and management of fatty acid oxidation disorders. Rev. Endocr. Metab. Disord. 2020, 21, 479–493. [Google Scholar] [CrossRef]
- Pietrzik, K.; Bronstrup, A. Vitamins B12, B6 and folate as determinants of homocysteine concentration in the healthy population. Eur. J. Pediatr. 1998, 157 (Suppl. S2), S135–S138. [Google Scholar] [CrossRef] [PubMed]
- Recommended Dietary Allowances, 10th ed.; The National Academies Collection: Reports Funded by National Institutes of Health; National Research Council-National Academy Press: Washington, DC, USA, 1989.
- National Institutes of Health. Vitamin A and Carotenoids. Available online: https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Thiamin. Available online: https://ods.od.nih.gov/factsheets/Thiamin-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Riboflavin. Available online: https://ods.od.nih.gov/factsheets/Riboflavin-HealthProfes-sional/ (accessed on 24 November 2023).
- National Institutes of Health. Niacin. Available online: https://ods.od.nih.gov/factsheets/Niacin-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Pantothenic Acid. Available online: https://ods.od.nih.gov/factsheets/PantothenicAc-id-HealthProfes-sional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin B6. Available online: https://ods.od.nih.gov/factsheets/VitaminB6-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin B7. Available online: https://ods.od.nih.gov/factsheets/Biotin-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Folate. Available online: https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin B12. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin C. Available online: https://ods.od.nih.gov/factsheets/VitaminC-HealthProfes-sional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin D. Available online: https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin E. Available online: https://ods.od.nih.gov/factsheets/VitaminE-HealthProfes-sional/ (accessed on 24 November 2023).
- National Institutes of Health. Vitamin K. Available online: https://ods.od.nih.gov/factsheets/VitaminK-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Copper. Available online: https://ods.od.nih.gov/factsheets/Copper-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Iron. Available online: https://ods.od.nih.gov/factsheets/Iron-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Selenium. Available online: https://ods.od.nih.gov/factsheets/Selenium-HealthProfessional/ (accessed on 24 November 2023).
- National Institutes of Health. Zinc. Available online: https://ods.od.nih.gov/factsheets/Zinc-HealthProfessional/ (accessed on 24 November 2023).
- Muhamad, R.; Akrivaki, A.; Papagiannopoulou, G.; Zavridis, P.; Zis, P. The Role of Vitamin B6 in Peripheral Neuropathy: A Systematic Review. Nutrients 2023, 15, 2823. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Feldman, H.H.; Zhang, S.; Flowers, S.A.; Luchsinger, J.A. Pharmacological thiamine levels as a therapeutic approach in Alzheimer’s disease. Front. Med. 2022, 9, 1033272. [Google Scholar] [CrossRef]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef]
- Bunik, V. The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot-Marie-Tooth Disease with the Compromised Status of Vitamin-Dependent Processes. Biology 2023, 12, 897. [Google Scholar] [CrossRef]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tong, Y.; Xiao, Y.; Huang, S.; Zhao, T.; Xia, X. Probiotic Bacillus subtilis contributes to the modulation of gut microbiota and blood metabolic profile of hosts. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2023, 272, 109712. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.L. Fat-Soluble Vitamins. Nurs. Clin. N. Am. 2021, 56, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Barragan, R.; Sanchez-Gonzalez, C.; Aranda, P.; Sorli, J.V.; Asensio, E.M.; Portoles, O.; Ortega-Azorin, C.; Villamil, L.V.; Coltell, O.; Llopis, J.; et al. Single and Combined Associations of Plasma and Urine Essential Trace Elements (Zn, Cu, Se, and Mn) with Cardiovascular Risk Factors in a Mediterranean Population. Antioxid. 2022, 11, 1991. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.T.; Misra, S.R.; Hussain, M. Nutritional Aspects of Essential Trace Elements in Oral Health and Disease: An Extensive Review. Scientifica 2016, 2016, 5464373. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F. Selenium, an antioxidant nutrient. Nutr. Clin. Care Off. Publ. Tufts Univ. 2002, 5, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R. The antioxidant properties of zinc. J. Nutr. 2000, 130, 1447S–1454S. [Google Scholar] [CrossRef]
- Wong, M.M.H.; Chan, K.Y.; Lo, K. Manganese Exposure and Metabolic Syndrome: A Systematic Review and Meta-Analysis. Nutrients 2022, 14, 825. [Google Scholar] [CrossRef]
- McWhorter, N.; Ndugga-Kabuye, M.K.; Puurunen, M.; Ernst, S.L. Complications of the Low Phenylalanine Diet for Patients with Phenylketonuria and the Benefits of Increased Natural Protein. Nutrients 2022, 14, 4960. [Google Scholar] [CrossRef]
- Baumgartner, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grunewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. International clinical guideline for the management of classical galactosemia: Diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 2017, 40, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Alcalde, C.; Belanger-Quintana, A.; Canedo-Villarroya, E.; Ceberio, L.; Chumillas-Calzada, S.; Correcher, P.; Couce, M.L.; Garcia-Arenas, D.; Gomez, I.; et al. Vitamin C and folate status in hereditary fructose intolerance. Eur. J. Clin. Nutr. 2022, 76, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Goldstein, J.; Austin, S.L.; Arn, P.; Bachrach, B.; Bali, D.S.; Chung, W.K.; El-Gharbawy, A.; Brown, L.M.; Kahler, S.; et al. Diagnosis and management of glycogen storage diseases type VI and IX: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 772–789. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, e1. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Helen Cross, J.; Dahlin, M.G.; et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Magrath, G.; MacDonald, A.; Whitehouse, W. Dietary practices and use of the ketogenic diet in the UK. Seizure 2000, 9, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Lammardo, A.M.; Robert, M.; Rocha, J.C.; van Rijn, M.; Ahring, K.; Belanger-Quintana, A.; MacDonald, A.; Dokoupil, K.; Ozel, H.G.; Goyens, P.; et al. Main issues in micronutrient supplementation in phenylketonuria. Mol. Genet. Metab. 2013, 110, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.H.; Rohr, F.; Frazier, D.; Cunningham, A.; Mofidi, S.; Ogata, B.; Splett, P.L.; Moseley, K.; Huntington, K.; Acosta, P.B.; et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 121–131. [Google Scholar] [CrossRef]
- Burlina, A.; Leuzzi, V.; Spada, M.; Carbone, M.T.; Paci, S.; Tummolo, A. The management of phenylketonuria in adult patients in Italy: A survey of six specialist metabolic centers. Curr. Med. Res. Opin. 2021, 37, 411–421. [Google Scholar] [CrossRef]
- Hanley, W.B.; Feigenbaum, A.; Clarke, J.T.; Schoonheyt, W.; Austin, V. Vitamin B12 deficiency in adolescents and young adults with phenylketonuria. Lancet 1993, 342, 997. [Google Scholar] [CrossRef]
- Crujeiras, V.; Aldamiz-Echevarria, L.; Dalmau, J.; Vitoria, I.; Andrade, F.; Roca, I.; Leis, R.; Fernandez-Marmiesse, A.; Couce, M.L. Vitamin and mineral status in patients with hyperphenylalaninemia. Mol. Genet. Metab. 2015, 115, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Barretto, J.R.; Silva, L.R.; Leite, M.E.; Boa-Sorte, N.; Pimentel, H.; Purificacao, A.C.; Carvalho, G.; Fontes, M.I.; Amorim, T. Poor zinc and selenium status in phenylketonuric children and adolescents in Brazil. Nutr. Res. 2008, 28, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, D.; Jarkovsky, J.; Vinohradska, H.; Konecna, P.; Machacova, L.; Dolezel, Z. Controlled diet in phenylketonuria and hyperphenylalaninemia may cause serum selenium deficiency in adult patients: The Czech experience. Biol. Trace Elem. Res. 2013, 154, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Miranda da Cruz, B.D.; Seidler, H.; Widhalm, K. Iron status and iron supplementation in children with classical phenylketonuria. J. Am. Coll. Nutr. 1993, 12, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Vugteveen, I.; Hoeksma, M.; Monsen, A.L.; Fokkema, M.R.; Reijngoud, D.J.; van Rijn, M.; van Spronsen, F.J. Serum vitamin B12 concentrations within reference values do not exclude functional vitamin B12 deficiency in PKU patients of various ages. Mol. Genet. Metab. 2011, 102, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.; Daly, A.; MacDonald, J.; Preece, M.A.; Santra, S.; Vijay, S.; Chakrapani, A.; MacDonald, A. The micronutrient status of patients with phenylketonuria on dietary treatment: An ongoing challenge. Ann. Nutr. Metab. 2014, 65, 42–48. [Google Scholar] [CrossRef]
- De Almeida, B.N.F.; Laufer, J.A.; Mezzomo, T.R.; Shimada, N.C.; Furtado, I.H.F.; Dias, M.; Pereira, R.M. Nutritional and metabolic parameters of children and adolescents with phenylketonuria. Clin. Nutr. ESPEN 2020, 37, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Kose, E.; Arslan, N. Vitamin/mineral and micronutrient status in patients with classical phenylketonuria. Clin. Nutr. 2019, 38, 197–203. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Sohrab, G.; Alaei, M.; Eini-Zinab, H.; Mohammadpour-Ahranjani, B.; Rastgoo, S.; Namkhah, Z. Growth and Nutritional Status of Phenylketonuric Children and Adolescents. BMC Pediatr. 2022, 22, 664. [Google Scholar] [CrossRef]
- Demirdas, S.; van Spronsen, F.J.; Hollak, C.E.M.; van der Lee, J.H.; Bisschop, P.H.; Vaz, F.M.; Ter Horst, N.M.; Rubio-Gozalbo, M.E.; Bosch, A.M. Micronutrients, Essential Fatty Acids and Bone Health in Phenylketonuria. Ann. Nutr. Metab. 2017, 70, 111–121. [Google Scholar] [CrossRef]
- Wiig, I.; Motzfeldt, K.; Loken, E.B.; Kase, B.F. Nutritional Consequences of Adhering to a Low Phenylalanine Diet for Late-Treated Adults with PKU: Low Phe Diet for Adults with PKU. JIMD Rep. 2013, 7, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Stolen, L.H.; Lilje, R.; Jorgensen, J.V.; Bliksrud, Y.T.; Almaas, R. High dietary folic Acid and high plasma folate in children and adults with phenylketonuria. JIMD Rep. 2014, 13, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Green, B.; Browne, R.; Firman, S.; Hill, M.; Rahman, Y.; Kaalund Hansen, K.; Adam, S.; Skeath, R.; Hallam, P.; Herlihy, I.; et al. Nutritional and Metabolic Characteristics of UK Adult Phenylketonuria Patients with Varying Dietary Adherence. Nutrients 2019, 11, 2459. [Google Scholar] [CrossRef] [PubMed]
- Kharitonchik, L.A.; Kodentsova, V.M.; Vrzhesinskaia, O.A.; Denisova, S.N.; Spirichev, V.B. Vitamin B 6 metabolism in phenylketonuria. Vopr. Meditsinskoi Khimii 2000, 46, 81–88. [Google Scholar]
- Kastrikina, L.N.; Kopylova, N.V.; Rybakova, E.P.; Ladodo, K.S.; Churdaleva, E.V.; Spirichev, V.B. The activity of glutathione-reductase and FAD-effect as indicators of the riboflavin level in experiments and in patients with phenylketonuria. Vopr. Pitan. 1975, 5, 12–18. [Google Scholar]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, O.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Tummolo, A.; Leone, P.; Tolomeo, M.; Solito, R.; Mattiuzzo, M.; Lepri, F.R.; Lore, T.; Cardinali, R.; De Giovanni, D.; Simonetti, S.; et al. Combined isobutyryl-CoA and multiple acyl-CoA dehydrogenase deficiency in a boy with altered riboflavin homeostasis. JIMD Rep. 2022, 63, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Touati, G.; Valayannopoulos, V.; Mention, K.; de Lonlay, P.; Jouvet, P.; Depondt, E.; Assoun, M.; Souberbielle, J.C.; Rabier, D.; Ogier de Baulny, H.; et al. Methylmalonic and propionic acidurias: Management without or with a few supplements of specific amino acid mixture. J. Inherit. Metab. Dis. 2006, 29, 288–298. [Google Scholar] [CrossRef]
- Frazier, D.M.; Allgeier, C.; Homer, C.; Marriage, B.J.; Ogata, B.; Rohr, F.; Splett, P.L.; Stembridge, A.; Singh, R.H. Nutrition management guideline for maple syrup urine disease: An evidence- and consensus-based approach. Mol. Genet. Metab. 2014, 112, 210–217. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Gass, J.M.; Vairo, F.P.E.; Farnham, K.M.; Atwal, H.K.; Macklin, S.; Klee, E.W.; Atwal, P.S. Maple syrup urine disease: Mechanisms and management. Appl. Clin. Genet. 2017, 10, 57–66. [Google Scholar] [CrossRef]
- Yannicelli, S.; Acosta, P.B.; Velazquez, A.; Bock, H.G.; Marriage, B.; Kurczynski, T.W.; Miller, M.; Korson, M.; Steiner, R.D.; Rutledge, L.; et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol. Genet. Metab. 2003, 80, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Pinar-Sueiro, S.; Martinez-Fernandez, R.; Lage-Medina, S.; Aldamiz-Echevarria, L.; Vecino, E. Optic neuropathy in methylmalonic acidemia: The role of neuroprotection. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S199–S203. [Google Scholar] [CrossRef] [PubMed]
- Traber, G.; Baumgartner, M.R.; Schwarz, U.; Pangalu, A.; Donath, M.Y.; Landau, K. Subacute bilateral visual loss in methylmalonic acidemia. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2011, 31, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Jurecki, E.; Ueda, K.; Frazier, D.; Rohr, F.; Thompson, A.; Hussa, C.; Obernolte, L.; Reineking, B.; Roberts, A.M.; Yannicelli, S.; et al. Nutrition management guideline for propionic acidemia: An evidence- and consensus-based approach. Mol. Genet. Metab. 2019, 126, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Haberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Steiner, R.D. Long-term management of patients with urea cycle disorders. J. Pediatr. 2001, 138, S56–S60, discussion S60–S51. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.H. Nutrition menagement of patients with inherited disorders of urea cycle enzymes. In Nutrition Menagement of Patients with Inherited Metabolic Disorders; Acosta, P.B., Ed.; Jones and Bartlett Publishers Inc.: Sudbury, MA, USA, 2009. [Google Scholar]
- Francini-Pesenti, F.; Gugelmo, G.; Lenzini, L.; Vitturi, N. Nutrient Intake and Nutritional Status in Adult Patients with Inherited Metabolic Diseases Treated with Low-Protein Diets: A Review on Urea Cycle Disorders and Branched Chain Organic Acidemias. Nutrients 2020, 12, 3331. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Champion, H.; Daly, A.; Dawson, S.; Dixon, M.; Dunlop, C.; Eardley, J.; Evans, S.; Ferguson, C.; Jankowski, C.; et al. Dietary management of urea cycle disorders: UK practice. J. Hum. Nutr. Diet. Off. J. Br. Diet. Assoc. 2012, 25, 398–404. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Gregersen, N.; Andresen, B.S.; Pedersen, C.B.; Olsen, R.K.; Corydon, T.J.; Bross, P. Mitochondrial fatty acid oxidation defects--remaining challenges. J. Inherit. Metab. Dis. 2008, 31, 643–657. [Google Scholar] [CrossRef]
- Merritt, J.L., 2nd; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef] [PubMed]
- Van Calcar, S.C.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.U.; Pendyal, S.; Wallace, L.S.; Hansen, J.G.; Stembridge, A.; et al. Nutrition management guideline for very-long chain acyl-CoA dehydrogenase deficiency (VLCAD): An evidence- and consensus-based approach. Mol. Genet. Metab. 2020, 131, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Goresky, C.A.; Bach, G.G.; Nadeau, B.E. On the uptake of materials by the intact liver. The transport and net removal of galactose. J. Clin. Investig. 1973, 52, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Succoio, M.; Sacchettini, R.; Rossi, A.; Parenti, G.; Ruoppolo, M. Galactosemia: Biochemistry, Molecular Genetics, Newborn Screening, and Treatment. Biomolecules 2022, 12, 968. [Google Scholar] [CrossRef]
- Ahmad, U.; Sharma, J. Fructose-1-Phosphate Aldolase Deficiency. In StatPearls; Ineligible Companies. Disclosure: Jyotsna Sharma Declares No Relevant Financial Relationships with Ineligible Companies; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gaughan, S.; Ayres, L.; Baker, P.R., II. Hereditary Fructose Intolerance. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chen, Y.T. Glycogen storage diseases. In The Metabolic & Molecular Basis of Inherited Diseases; Scriver, C.R., Beaudet, A.L., Sly, W.S., Vale, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1521–1552. [Google Scholar]
- Cori, G.T.; Cori, C.F. Glucose-6-phosphatase of the liver in glycogen storage disease. J. Biol. Chem. 1952, 199, 661–667. [Google Scholar] [CrossRef]
- Gierke, E.V. Hepato-nephro-megalia-glycogenica (Glykogenspeicherkrankheit der Leber und Nieren). Beitr. Pathol. Anat. 1929, 82, 497–513. [Google Scholar]
- Christodoulides, S.S.; Neal, E.G.; Fitzsimmons, G.; Chaffe, H.M.; Jeanes, Y.M.; Aitkenhead, H.; Cross, J.H. The effect of the classical and medium chain triglyceride ketogenic diet on vitamin and mineral levels. J. Hum. Nutr. Diet. Off. J. Br. Diet. Assoc. 2012, 25, 16–26. [Google Scholar] [CrossRef]
- Scholl-Burgi, S.; Holler, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef]
- Mereis, M.; Wanders, R.J.A.; Schoonen, M.; Dercksen, M.; Smuts, I.; van der Westhuizen, F.H. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int. J. Biochem. Cell Biol. 2021, 132, 105899. [Google Scholar] [CrossRef]
- Bergqvist, A.G.; Schall, J.I.; Stallings, V.A. Vitamin D status in children with intractable epilepsy, and impact of the ketogenic diet. Epilepsia 2007, 48, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Willmott, N.S.; Bryan, R.A. Case report: Scurvy in an epileptic child on a ketogenic diet with oral complications. Eur. Arch. Paediatr. Dent. Off. J. Eur. Acad. Paediatr. Dent. 2008, 9, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.S.; Billson, F.A. Optic neuropathy in ketogenic diet. Br. J. Ophthalmol. 1979, 63, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Zupec-Kania, B.; Zupanc, M.L. Long-term management of the ketogenic diet: Seizure monitoring, nutrition, and supplementation. Epilepsia 2008, 49 (Suppl. S8), 23–26. [Google Scholar] [CrossRef] [PubMed]
- Zupec-Kania, B.A.; Spellman, E. An overview of the ketogenic diet for pediatric epilepsy. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2008, 23, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Williams, S.; Basualdo-Hammond, C.; Stephens, D.; Curtis, R. A prospective study: Growth and nutritional status of children treated with the ketogenic diet. J. Am. Diet. Assoc. 2003, 103, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Tragni, V.; Primiano, G.; Tummolo, A.; Cafferati Beltrame, L.; La Piana, G.; Sgobba, M.N.; Cavalluzzi, M.M.; Paterno, G.; Gorgoglione, R.; Volpicella, M.; et al. Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs. Molecules 2022, 27, 3494. [Google Scholar] [CrossRef]
- Bakaloudi, D.R.; Halloran, A.; Rippin, H.L.; Oikonomidou, A.C.; Dardavesis, T.I.; Williams, J.; Wickramasinghe, K.; Breda, J.; Chourdakis, M. Intake and adequacy of the vegan diet. A systematic review of the evidence. Clin. Nutr. 2021, 40, 3503–3521. [Google Scholar] [CrossRef]
- Evans, M.; Truby, H.; Boneh, A. The relationship between dietary intake, growth and body composition in Phenylketonuria. Mol. Genet. Metab. 2017, 122, 36–42. [Google Scholar] [CrossRef]
- Stroup, B.M.; Ney, D.M.; Murali, S.G.; Rohr, F.; Gleason, S.T.; van Calcar, S.C.; Levy, H.L. Metabolomic Insights into the Nutritional Status of Adults and Adolescents with Phenylketonuria Consuming a Low-Phenylalanine Diet in Combination with Amino Acid and Glycomacropeptide Medical Foods. J. Nutr. Metab. 2017, 2017, 6859820. [Google Scholar] [CrossRef]
- Youness, R.A.; Dawoud, A.; ElTahtawy, O.; Farag, M.A. Fat-soluble vitamins: Updated review of their role and orchestration in human nutrition throughout life cycle with sex differences. Nutr. Metab. 2022, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Swerdlow, R.H.; Inzitari, M.; Olaso-Gonzalez, G.; Vina, J. Multimodal strategy to rescue the brain in mild cognitive impairment: Ketogenic oral nutrition supplementation with B vitamins and aerobic exercise. Eur. J. Clin. Investig. 2022, 52, e13806. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Hossain, M.; Sanin, K.I. Global burden of maternal and child undernutrition and micronutrient deficiencies. Ann. Nutr. Metab. 2012, 61 (Suppl. S1), 8–17. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.; Evans, S.; Pinto, A.; Ashmore, C.; MacDonald, A. Protein Substitutes in PKU.; Their Historical Evolution. Nutrients 2021, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Impact of pregnancy on inborn errors of metabolism. Rev. Endocr. Metab. Disord. 2018, 19, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Manta-Vogli, P.D.; Schulpis, K.H.; Dotsikas, Y.; Loukas, Y.L. Nutrition and medical support during pregnancy and lactation in women with inborn errors of intermediary metabolism disorders (IEMDs). J. Pediatr. Endocrinol. Metab. JPEM 2020, 33, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.A.; Kenney, M.K.; Harris, K.B.; Singh, R.H.; Cameron, C.A.; Kraszewski, J.N.; Levy-Fisch, J.; Shuger, J.F.; Greene, C.L.; Lloyd-Puryear, M.A.; et al. Insurance coverage of medical foods for treatment of inherited metabolic disorders. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 978–982. [Google Scholar] [CrossRef]
- Verduci, E.; Moretti, F.; Bassanini, G.; Banderali, G.; Rovelli, V.; Casiraghi, M.C.; Morace, G.; Borgo, F.; Borghi, E. Phenylketonuric diet negatively impacts on butyrate production. Nutr. Metab. Cardiovasc. Dis. NMCD 2018, 28, 385–392. [Google Scholar] [CrossRef]
- Colonetti, K.; Bento Dos Santos, B.; Nalin, T.; Moura de Souza, C.F.; Triplett, E.W.; Dobbler, P.T.; Schwartz, I.V.D.; Roesch, L.F.W. Hepatic glycogen storage diseases are associated to microbial dysbiosis. PLoS ONE 2019, 14, e0214582. [Google Scholar] [CrossRef]
- Montanari, C.; Parolisi, S.; Borghi, E.; Putignani, L.; Bassanini, G.; Zuvadelli, J.; Bonfanti, C.; Tummolo, A.; Dionisi Vici, C.; Biasucci, G.; et al. Dysbiosis, Host Metabolism, and Non-communicable Diseases: Trialogue in the Inborn Errors of Metabolism. Front. Physiol. 2021, 12, 716520. [Google Scholar] [CrossRef]


{kind=link}
| Micronutrient | Main Function | RDA/DRI/AI | Dose of Supplementation Recommended | Nutritional Sources | Biomarkers | Deficiency Clinical Manifestation | Web Ref. |
|---|---|---|---|---|---|---|---|
| Vitamin A (retinoids and carotenoids) | Regeneration of visual pigment, maintenance of mucosal membranes, and immune function | 300–900 mcg RAE/day | Rarely recommended | Some types of fish, such as herring and salmon. Beef liver and other organ meats. Green leafy vegetables and other green, orange, and yellow vegetables such as spinach, sweet potatoes, carrots, broccoli, and winter squash. Fruits, including cantaloupe, mangos, and apricots. Dairy products, such as milk and cheese. Fortified breakfast cereals, eggs | Serum retinol and retinyl esters in serum | Clinical manifestations are represented by the gradual development of night blindness, an increased frequency of infections, the development of xeroderma and follicular hyperkeratosis, xerophthalmia, and conjunctival xerosis | [19] |
| Vitamin B1 (thiamin) | Carbohydrate metabolism, ATP production | 0.2–1.2 mg/day | 3–5 mg/day up to 10 mg/day in severe deficiencies | Whole grains and fortified bread, cereal, pasta, and rice. Meat (especially pork) and fish. Legumes, seeds, and nuts | Red blood cells (RBC) or whole blood thiamine diphosphate (ThDP) | In its early stage, thiamine deficiency can cause weight loss, anorexia, confusion, short-term memory loss, muscle weakness, and cardiovascular symptoms. The most common effect is beriberi, characterized by peripheral neuropathy and wasting, which can lead to impaired sensory, motor, and reflex functions. Another common manifestation is Wernicke–Korsakoff syndrome, i.e., Wernicke’s encephalitis, characterized by peripheral neuropathy, and Korsakoff’s psychosis. | [20] |
| Vitamin B2 (riboflavin, vitamin G) | Essential component of 2 coenzymes, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), involved in energy production, cellular function, growth, and development, and metabolism of fats, drugs, and steroids | 0.3–1.3 mg/day | 5–10 mg/day up to 160 mg/day in severe deficiencies | Eggs, organ meats (such as kidneys and liver), lean meats, and low-fat milk. Some vegetables (such as mushrooms and spinach). Fortified cereals, bread, and grain products | Erythrocyte glutathione reductase activity test | Deficiency is manifested by oral buccal lesions (cheilosis, glossitis, and angular stomatitis) and seborrheic dermatitis of the face, trunk, and scrotum. Other manifestations are ocular itching, burning, dryness, corneal inflammation, and photophobia, and normochromic, normocytic anemia and marrow aplasia | [21] |
| Vitamin B3 (niacin, nicotinic acid, vitamin PP) | Converted into its main metabolically active form, the coenzyme NAD. It is involved in redox reactions | 2–16 NEs/day | 250–500 mg/day | Animal foods, such as poultry, beef, pork, and fish. Some types of nuts, legumes, and grains. Enriched and fortified foods, such as many breads and cereals | Urinary determination of the two major niacin metabolites, N-methyl-nicotinamide (NMN) and N-methyl-2-pyridone-carboxamide (2-Pyr), is used to determine niacin biomarker status | Diarrhea, dermatitis, and dementia, collectively known as “pellagra” or “the three D disease”, and even death (four D) if not recognized and treated promptly | [22] |
| Vitamin B5 (pantothenic acid) | Used for the synthesis of coenzyme A (CoA) and the citric acid cycle | 1.7–5 mg/day | 10 mg up to 1000 mg/day | Beef, poultry, seafood, and organ meats, eggs and milk, vegetables such as mushrooms (especially shiitakes), avocados, potatoes, and broccoli. Whole grains, such as whole wheat, brown rice, and oats, peanuts, sunflower seeds, and chickpeas | Whole blood and urine (24 h collection) are the sample matrices that have proven to be the most informative | Headache Fatigue Irritability, restlessness Disturbed sleep Nausea, vomiting, stomach cramps Numbness or burning sensation in hands or feet Muscle cramps | [23] |
| Vitamin B6 (pyridoxin) | In its active forms, pyridoxal 5′ phosphate (PLP) and pyridoxamine 5′ phosphate (PMP), it is involved in > 100 reactions, related to protein metabolism, carbohydrates, and lipids, biosynthesis of neurotransmitters and in maintaining normal levels of homocysteine; gluconeogenesis and glycogenolysis, immune function, hemoglobin | 0.1–1.7 mg/day | 6–50 mg/day up to 200 mg/day in severe deficiencies | Poultry, fish, and organ meats, potatoes and other starchy vegetables | Plasma levels of PLP correlate with pyridoxine intake and body stores and are recognized as a status biomarker | Microcytic anemia Skin conditions (seborrheic dermatitis with cheilosis and glossitis) Depression Confusion Lowered immunity, angular stomatitis | [24] |
| Vitamin B7 (biotin or vitamin H) | Coenzyme for five carboxylase enzymes, which are involved in the digestion of carbohydrates, synthesis of fatty acids, and gluconeogenesis | 5–30 mcg/day | 5–10 mg/day | Meat, fish, eggs, and organ meats (such as liver), seeds and nuts, certain vegetables (such as sweet potatoes, spinach, and broccoli) | Direct analysis of biotin in blood, serum/plasma, and urine (MS/MS) Indirect measurement: urinary excretion (24 h urine) of biotin and of metabolites produced by biotin-dependent carboxylases and related metabolic pathways (3-hydroxyisovaleric acid, 3-hydroxyisovalerylcarnitine). 3-hydroxyisovaleric acid and 3- Hydroxyisovalerylcarnitine biotinidase activity | Thinning hair Scaly skin rashes around eyes, nose, mouth Brittle nails | [25] |
| Vitamin B9 (folic acid) | Folate functions as a coenzyme or cosubstrate in single-carbon transfers in the synthesis of nucleic acids (DNA and RNA) and metabolism of amino acids. It plays the most important role in the conversion of homocysteine to methionine in the synthesis of S-adenosyl-methionine, in the methylation of deoxyuridylate to thymidylate, in the formation of DNA, and is required for proper cell division. | 65–400 mcg DFE/day | 1–5 mg/day | Beef liver, vegetables (especially asparagus, brussels sprouts, and dark green leafy vegetables such as spinach and mustard greens), fruits and fruit juices (especially oranges and orange juice), nuts, beans, and peas (such as peanuts, black-eyed peas, and kidney beans) | Levels of folate in serum/plasma or RBC | Most symptoms of folate deficiency overlap with those of cobalamin deficiency, i.e., megaloblastic anemia and pancytopenia, glossitis, angular stomatitis, oral ulcers, neuropsychiatric manifestations, including depression, irritability, insomnia, cognitive impairment, psychosis, anorexia, and fatigue | [26] |
| Vitamin B12 (cobalamin) | Vitamin B12 is required for the development, myelination, and function of the central nervous system, red blood cell formation, and DNA synthesis. It is a cofactor for two enzymes, methionine synthase and L-methylmalonyl-CoA mutase | 0.4–2.4 mcg/day | 100–1000 mcg IM/SC | Fish, meat, poultry, eggs, milk, and other dairy products. Clams and beef liver, enriched breakfast cereals, nutritional yeasts | Direct cobalamin levels measurement | Megaloblastic anemia—a condition of larger-than-normal-sized red blood cells and a smaller-than-normal amount; this occurs because there is not enough vitamin B12 in the diet or poor absorption Pernicious anemia—a type of megaloblastic anemia caused by a lack of intrinsic factor so that vitamin B12 is not absorbed Fatigue, weakness Nerve damage with numbness, tingling in the hands and legs Memory loss, confusion Dementia Depression Seizures | [27] |
| Vitamin C (ascorbic acid and ascorbates) | It is required for the biosynthesis of collagen, L-carnitine, and certain neurotransmitters. It is also involved in protein metabolism and it is an important physiological antioxidant. It plays an important role in immune function and improves the absorption of nonheme iron, the form of iron present in plant-based foods | 15–90 mg/day | 200 mg up to 2–3 g/day | Citrus fruits (such as oranges and grapefruit) and their juices, as well as red and green pepper and kiwifruit. Broccoli, strawberries, cantaloupe, baked potatoes, and tomatoes. Fortified foods and beverages | Assessment of vitamin C status can be determined from its concentration in either plasma or leukocytes | Scurvy, the hallmark disease of severe vitamin C deficiency, displays symptoms resulting from the loss of collagen that weakens connective tissues: skin spots caused by bleeding and bruising from broken blood vessels Swelling or bleeding of gums, and eventual loss of teeth Hair loss Delayed healing of skin wounds Fatigue, malaise Iron-deficiency anemia due to decreased absorption of non-heme iron | [28] |
| Vitamin D (cholecalciferol, ergocalciferol) | It is important for building and maintaining healthy bones and teeth. It reduces inflammation. It aids in the process of cell growth. It plays a role in immune function and supports muscle function and strength | 10–20 mcg-day | 400–1000 IU/day (infants) up to more than 10,000 IU/day (high-risk adults) | Butter and fatty cheeses, beef liver. Fortified milk, soy milk, oat milk, and almond milk. Fatty fish (like trout, salmon, tuna, and mackerel) and fish liver oils. Fortified food | Serum/plasma concentrations of total 25-hydroxyvitamin D (25-OHD), the sum of 25-OHD3 and 25-OHD2, are recognized as a valid biomarker for vitamin D status | Rickets Osteomalacia | [29] |
| Vitamin E (tocopherol and tocotrienols) | Antioxidants protect cells from the damaging effects of free radicals, which are molecules that contain an unshared electron. Free radicals damage cells and might contribute to the development of cardiovascular disease and cancer. Unshared electrons are highly energetic and react rapidly with oxygen to form reactive oxygen species | 4–15 mg/day | 15–25 mg/kg or mixed tocopherols 200 IU/day | Vegetable oils like wheat germ, sunflower, and safflower oils, corn and soybean oils. Nuts and seeds. Green vegetables, such as spinach and broccoli. Fortified food | Vitamin E status is determined by the quantification of tocopherol in blood plasma or serum | Retinopathy (damage to the retina of the eyes that can impair vision) Peripheral neuropathy (damage to the peripheral nerves, usually in the hands or feet, causing weakness or pain) Ataxia (loss of control of body movements) Decreased immune function | [30] |
| Vitamin K (vitamin K1 and K2) | It functions as a coenzyme required for the protein synthesis. It is involved in blood clotting, hemostasis, and bone metabolism | 2–120 mcg/day | 1–2 mg/day (infant) 90–120 mcg/day (adult) | Green leafy vegetables, such as spinach, kale, broccoli, and lettuce. Vegetable oils. Some fruits, such as blueberries and figs. Meat, cheese, eggs, and soybeans | The quantification of circulating phylloquinone (vitamin K1) in blood plasma or serum remains the most commonly used marker of vitamin K status, although it is mainly an indirect biomarker of short-term phylloquinone intake. | A longer time for blood to clot or a prolonged prothrombin time (as measured in a physician’s office) Bleeding Hemorrhaging Osteopenia or osteoporosis | [31] |
| Copper | It is required for adequate growth, cardiovascular integrity, lung elasticity, neovascularization, neuroendocrine function, and iron metabolism | 200–900 mcg/day | 4–8 mg/day | Beef liver and shellfish such as oysters. Nuts, seeds, and chocolate. Wheat-bran cereals and whole-grain products, potatoes, mushrooms, avocados, chickpeas, and tofu | Ceruloplasmin: 98% of circulating copper is bound to ceruloplasmin | Microcytic anemia, neutropenia, osteoporosis, and hair depigmentation (copper is essential for melanin synthesis) | [32] |
| Iron | It is involved in oxygen and lipid metabolism, in protein production, cellular respiration, and DNA synthesis | 0.27–18 mg/day | 150–200 mg/day | Lean meat, seafood, and poultry. Iron-fortified breakfast cereals and breads. White beans, lentils, spinach, kidney beans, and peas. Nuts and some dried fruits, such as raisins | Plasma iron, hemoglobin, mean red cell volume (MCV), transferrin, transferrin saturation, total iron binding capacity (TIBC), hepcidin, ferritin Bone marrow iron soluble transferrin receptor | Microcytic anemia and/or low ferritin levels | [33] |
| Selenium | It is an active immunomodulator and antioxidant. It takes part in thyroxine conversion to triiodethyronine in thyroid hormone biosynthesis. As a sperm antioxidant, it protects its motility and fertility. Selenium is a serious factor in the biological and antioxidant protection of vascular endothelium, of low-density lipoproteins, protection of DNA, chromosomes | 15–55 mcg/day | 60–100 mg/day | Seafood, meat, poultry, eggs, and dairy products. Breads, cereals, and other grain products | Whole blood or plasma/serum selenium concentration | Increased incidence and virulence of viral infections, cardiac and skeletal muscle myopathy, and skin and nail effects (selenium concentration <0.4 mmol/L (<32 mg/L)) | [34] |
| Zinc | Structural, catalytic, and intracellular and intercellular signaling component | 2–11 mg/die | 20–40 mg/day | Oysters, meat, fish, poultry, seafood such as crab and lobsters, and fortified breakfast cereals. Beans, nuts, whole grains, eggs, and dairy products | Whole blood, plasma, serum, urine | Alopecia, skin rash of face, groin, hands, and feet, growth retardation, delayed sexual development and bone maturation, impaired wound healing and immune function, diarrhea, and blunting of taste and smell | [35] |
| Category of Disorder | Type of Disorder | Diet Regimen | Principal Category of Limited Food | Ref. |
|---|---|---|---|---|
| Amino acid disorders | Phenylketonuria | Low Phenylalanine intake | Meat, fish, eggs, pulses, milk and dairy products, cereals | [13] |
| Organic acidosis (OA) | Low natural protein intake | Meat, fish, eggs, pulses, milk and dairy products, cereals | [14,49] | |
| UCDs | Low natural protein intake | Meat, fish, eggs, pulses, milk and dairy products, cereals | [15] | |
| Fatty acid oxidation disorders | VLCADD | Low intake of long-chain fatty acids | Full-fat and semi-skimmed milk, egg yolks, fatty fish and meat, cheese, butter, margarine, vegetable oil, dried fruit, oilseeds, chocolate, baked products, industrial products | [16] |
| Carbohydrate disorders | Galactosemia | Galactose-restricted diet | Milk and derivatives | [50] |
| Hereditary fructose intolerance | Minimal fructose and absolute exclusion of sucrose and alimentary additives like caramel (E150), sweeteners isomalt (E963), maltitol (E965) mannitol (E421), sorbitol (E420), xylitol (E967) intake | Fruit, honey, vegetables, other products containing sugar | [51] | |
| Glycogen storage disorders (I, III, VI, IX) | Fructose, sucrose, and galactose exclusion (I) Moderately high protein and low sugar intake (III) Low carbohydrate intake (VII) | Fruit, honey, vegetables, products containing sugar | [52,53] | |
| IMD treated with ketogenic diet | GLUT1 deficiency PDH deficiency | Low carbohydrates and high fat intake | Fruit, dessert pastry, sweets, juice, pasta, cereals and baked products, potatoes, pulses | [54,55] |
| Category of Disorders | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Amino acid disorders | Fatty acid oxidation disorders | Carbohydrate disorders | IMDs treated with KD | ||||||
| Type of disorder | |||||||||
| PKU | OAs | UCDs | VLCADD | Galactosemias | HF intolerance | GSDs (I, III, VI, IX) | GLUT1-D PDH-D | ||
| Vitamins | A | ||||||||
| B1 | |||||||||
| B2 | |||||||||
| B3 | |||||||||
| B5 | |||||||||
| B6 | |||||||||
| B7 | |||||||||
| B9 | |||||||||
| B12 | |||||||||
| C | |||||||||
| D | |||||||||
| E | |||||||||
| K | |||||||||
| Minerals | Ca | ||||||||
| Cu | |||||||||
| K | |||||||||
| Fe | |||||||||
| I | |||||||||
| Mg | |||||||||
| Mn | |||||||||
| Se | |||||||||
| Zn | |||||||||
| High micronutrient level | |||||||||
| Low micronutrient level | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tummolo, A.; Carella, R.; De Giovanni, D.; Paterno, G.; Simonetti, S.; Tolomeo, M.; Leone, P.; Barile, M. Micronutrient Deficiency in Inherited Metabolic Disorders Requiring Diet Regimen: A Brief Critical Review. Int. J. Mol. Sci. 2023, 24, 17024. https://doi.org/10.3390/ijms242317024
Tummolo A, Carella R, De Giovanni D, Paterno G, Simonetti S, Tolomeo M, Leone P, Barile M. Micronutrient Deficiency in Inherited Metabolic Disorders Requiring Diet Regimen: A Brief Critical Review. International Journal of Molecular Sciences. 2023; 24(23):17024. https://doi.org/10.3390/ijms242317024
Chicago/Turabian StyleTummolo, Albina, Rosa Carella, Donatella De Giovanni, Giulia Paterno, Simonetta Simonetti, Maria Tolomeo, Piero Leone, and Maria Barile. 2023. "Micronutrient Deficiency in Inherited Metabolic Disorders Requiring Diet Regimen: A Brief Critical Review" International Journal of Molecular Sciences 24, no. 23: 17024. https://doi.org/10.3390/ijms242317024