
AsHC 360 Exposure Influence on Epileptiform Discharges in Hippocampus of Infantile Male Rats In Vitro

Abstract
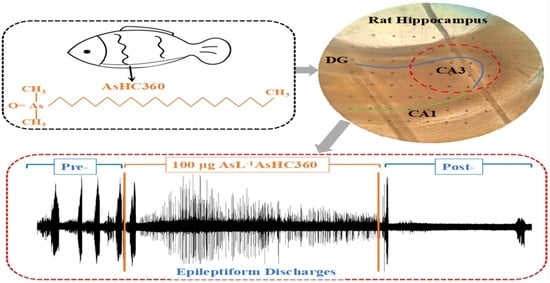
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Mg2+-Free ACSF Induces Normal EDs
2.2. The Effect of AsHC 360 Exposure on EDs
2.3. Agonist Modulates the Effects of AsHC 360 on EDs
2.4. Comparison of Concentration-Dependent Effects on EDs
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Standards
4.2. Animals and Hippocampal Slices Preparation
4.3. Epileptiform Activity Induction
4.4. Experimental Protocol
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oremland, R.S.; Stolz, J.F. The ecology of arsenic. Science 2003, 300, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Tolins, M.; Ruchirawat, M.; Landrigan, P. The developmental neurotoxicity of arsenic: Cognitive and behavioral consequences of early life exposure. Ann. Glob. Health 2014, 80, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Levitsky, D.O. Arsenolipids. Prog. Lipid Res. 2004, 43, 403–448. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, U.; Jakubik, S.; Kuballa, J.; Jantzen, E. Determination of Arsenic Species in Fish Oil after Acid. Digestion. Microchim. Acta 2005, 151, 249–255. [Google Scholar] [CrossRef]
- Lischka, S.; Arroyo-Abad, U.; Mattusch, J.; Kühn, A.; Piechotta, C. The high diversity of arsenolipids in herring fillet (Clupea harengus). Talanta 2013, 110, 144–152. [Google Scholar] [CrossRef]
- Rumpler, A.; Edmonds, J.S.; Katsu, M.; Jensen, K.B.; Goessler, W.; Raber, G.; Gunnlaugsdottir, H.; Francesconi, K.A. Arsenic-containing long-chain fatty acids in cod-liver oil: A result of biosynthetic infidelity? Angew. Chem. Int. Ed. Engl. 2008, 47, 2665–2667. [Google Scholar] [CrossRef]
- Sele, V.; Sloth, J.J.; Holmelid, B.; Valdersnes, S.; Skov, K.; Amlund, H. Arsenic-containing fatty acids and hydrocarbons in marine oils—Determination using reversed-phase HPLC-ICP-MS and HPLC-qTOF-MS. Talanta 2014, 121, 89–96. [Google Scholar] [CrossRef]
- Amayo, K.O.; Raab, A.; Krupp, E.M.; Marschall, T.; Horsfall, M., Jr.; Feldmann, J. Arsenolipids show different profiles in muscle tissues of four commercial fish species. J. Trace Elem. Med. Biol. 2014, 28, 131–137. [Google Scholar] [CrossRef]
- Meyer, S.; Matissek, M.; Müller, S.M.; Taleshi, M.S.; Ebert, F.; Francesconi, K.A.; Schwerdtle, T. In vitro toxicological characterisation of three arsenic-containing hydrocarbons. Metallomics 2014, 6, 1023–1033. [Google Scholar] [CrossRef]
- Meyer, S.; Schulz, J.; Jeibmann, A.; Taleshi, M.S.; Ebert, F.; Francesconi, K.A.; Schwerdtle, T. Arsenic-containing hydrocarbons are toxic in the in vivo model Drosophila melanogaster. Metallomics 2014, 6, 2010–2014. [Google Scholar] [CrossRef]
- Witt, B.; Ebert, F.; Meyer, S.; Francesconi, K.A.; Schwerdtle, T. Assessing neurodevelopmental effects of arsenolipids in pre-differentiated human neurons. Mol. Nutr. Food Res. 2017, 61, 1700199. [Google Scholar] [CrossRef] [PubMed]
- Witt, B.; Meyer, S.; Ebert, F.; Francesconi, K.A.; Schwerdtle, T. Toxicity of two classes of arsenolipids and their water-soluble metabolites in human differentiated neurons. Arch. Toxicol. 2017, 91, 3121–3134. [Google Scholar] [CrossRef] [PubMed]
- Amayo, K.O.; Raab, A.; Krupp, E.M.; Gunnlaugsdottir, H.; Feldmann, J. Novel identification of arsenolipids using chemical derivatizations in conjunction with RP-HPLC-ICPMS/ESMS. Anal. Chem. 2013, 85, 9321–9327. [Google Scholar] [CrossRef] [PubMed]
- Taleshi, M.S.; Jensen, K.B.; Raber, G.; Edmonds, J.S.; Gunnlaugsdottir, H.; Francesconi, K.A. Arsenic-containing hydrocarbons: Natural compounds in oil from the fish capelin, Mallotus villosus. Chem. Commun. 2008, 21, 4706–4707. [Google Scholar] [CrossRef]
- Zheng, Y.; Tian, C.X.; Dong, L.; Tian, L.; Glabonjat, R.A.; Xiong, C. Effect of arsenic-containing hydrocarbon on the long-term potentiation at Schaffer Collateral-CA1 synapses from infantile male rat. Neurotoxicology 2021, 84, 198–207. [Google Scholar] [CrossRef]
- Stiboller, M.; Raber, G.; Lenters, V.; Gjengedal, E.L.F.; Eggesbø, M.; Francesconi, K.A. Arsenolipids Detected in the Milk of Nursing Mothers. Environ. Sci. Technol. Lett. 2017, 4, 273–279. [Google Scholar] [CrossRef]
- Xiong, C.; Stiboller, M.; Glabonjat, R.A.; Rieger, J.; Paton, L.; Francesconi, K.A. Transport of arsenolipids to the milk of a nursing mother after consuming salmon fish. J. Trace Elem. Med. Bio 2020, 9, 126502. [Google Scholar] [CrossRef]
- Grandjean, P.; Landrigan, P.J. Developmental neurotoxicity of industrial chemicals. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef]
- Kim, M.; Seo, S.; Sung, K.; Kim, K. Arsenic exposure in drinking water alters the dopamine system in the brains of C57BL/6 mice. Biol. Trace Elem. Res. 2014, 162, 175–180. [Google Scholar] [CrossRef]
- Wang, X.; Mu, X.; Zhang, J.; Huang, Q.; Alamdar, A.; Tian, M.; Liu, L.; Shen, H. Serum metabolomics reveals that arsenic exposure disrupted lipid and amino acid metabolism in rats: A step forward in understanding chronic arsenic toxicity. Metallomics 2015, 7, 544–552. [Google Scholar] [CrossRef]
- Calatayud, M.; Barrios, J.A.; Vélez, D.; Devesa, V. In vitro study of transporters involved in intestinal absorption of inorganic arsenic. Chem. Res. Toxicol. 2012, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shen, J.; Carbrey, J.M.; Mukhopadhyay, R.; Agre, P.; Rosen, B.P. Arsenite transport by mammalian aquaglyceroporins AQP7 and AQP9. Proc. Natl. Acad. Sci. USA 2002, 99, 6053–6058. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Ishii, K.; Seto, Y.; Hosoya, T.; Tanaka, R.; Nakayama, T.; Iwasaki, N.; Shibata, Y.; Tamaoka, A. Long-term accumulation of diphenylarsinic acid in the central nervous system of cynomolgus monkeys. Arch. Toxicol. 2017, 91, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Peña, L.C.; Petrosyan, P.; Morales, M.; González, N.B.; Gutiérrez-Ospina, G.; Del Razo, L.M.; Gonsebatt, M.E. Arsenic species, AS3MT amount, and AS3MT gene expression in different brain regions of mouse exposed to arsenite. Environ. Res. 2010, 110, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Colom, L.V.; Saggau, P. Spontaneous interictal-like activity originates in multiple areas of the CA2-CA3 region of hippocampal slices. J. Neurophysiol. 1994, 71, 1574–1585. [Google Scholar] [CrossRef]
- Dai, Y.; Song, Y.; Xie, J.; Xu, S.; Li, X.; He, E.; Yin, H.; Cai, X. In Vivo Microelectrode Arrays for Detecting Multi-Region Epileptic Activities in the Hippocampus in the Latent Period of Rat Model of Temporal Lobe Epilepsy. Micromachines 2021, 12, 659. [Google Scholar] [CrossRef]
- Luo, J.; Qiu, Z.; Chen, J.; Zhang, L.; Liu, W.; Tan, Y.; Shu, W. Maternal and early life arsenite exposure impairs neurodevelopment and increases the expression of PSA-NCAM in hippocampus of rat offspring. Toxicology 2013, 311, 99–106. [Google Scholar] [CrossRef]
- Tyler, C.R.; Allan, A.M. The Effects of Arsenic Exposure on Neurological and Cognitive Dysfunction in Human and Rodent Studies: A Review. Curr. Environ. Health Rep. 2014, 1, 132–147. [Google Scholar] [CrossRef]
- Maekawa, F.; Tsuboi, T.; Oya, M.; Aung, K.H.; Tsukahara, S.; Pellerin, L.; Nohara, K. Effects of sodium arsenite on neurite outgrowth and glutamate AMPA receptor expression in mouse cortical neurons. Neurotoxicology 2013, 37, 197–206. [Google Scholar] [CrossRef]
- Nelson-Mora, J.; Escobar, M.L.; Rodríguez-Durán, L.; Massieu, L.; Montiel, T.; Rodríguez, V.M.; Hernández-Mercado, K.; Gonsebatt, M.E. Gestational exposure to inorganic arsenic (iAs3+) alters glutamate disposition in the mouse hippocampus and ionotropic glutamate receptor expression leading to memory impairment. Arch. Toxicol. 2018, 92, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, K.; Dong, L.; Tian, C. Study on the mechanism of high-frequency stimulation inhibiting low-Mg2+-induced epileptiform discharges in juvenile rat hippocampal slices. Brain Res. Bull. 2020, 165, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.M.; Ebert, F.; Raber, G.; Meyer, S.; Bornhorst, J.; Hüwel, S.; Galla, H.J.; Francesconi, K.A.; Schwerdtle, T. Effects of arsenolipids on in vitro blood-brain barrier model. Arch. Toxicol. 2018, 92, 823–832. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, W.; Gong, H.; Li, G.; Jiang, Q.; Liang, P.; Zheng, H.; Zhang, P. Low-intensity ultrasound suppresses low-Mg2+-induced epileptiform discharges in juvenile mouse hippocampal slices. J. Neural Eng. 2019, 16, 036006. [Google Scholar] [CrossRef] [PubMed]
- Qutub, A.A.; Hunt, C.A. Glucose transport to the brain: A systems model. Brain Res. Brain Res. Rev. 2005, 49, 595–617. [Google Scholar] [CrossRef]
- Lomako, J.; Lomako, W.M.; Whelan, W.J.; Dombro, R.S.; Neary, J.T.; Norenberg, M.D. Glycogen synthesis in the astrocyte: From glycogenin to proglycogen to glycogen. FASEB J. 1993, 7, 1386–1393. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, L.; Magistretti, J.P. Brain energy metabolism: Focuson astrocyte—Neuron m etabolic cooperation. CellMetab 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Koifman, J.; Shin, D.S.; Ye, H.; Florez, C.M.; Zhang, L.; Valiante, T.A.; Carlen, P.L. Transition to seizure: Ictal discharge is preceded by exhausted presynaptic GABA release in the hippocampal CA3 region. J. Neurosci. 2012, 32, 2499–2512. [Google Scholar] [CrossRef]
- Navarro, V.; Martinerie, J.; Quyen, M.L.V.; Clemenceau, S.; Adam, C.; Baulac, M.; Varela, F. Seizure anticipation in human neocortical partial epilepsy. Brain 2002, 125, 640–655. [Google Scholar] [CrossRef]
- Slone, E.; Westwood, E.; Dhaliwal, H.; Federico, P.; Dunn, J.F. Near-infrared spectroscopy shows preictal haemodynamic changes in temporal lobe epilepsy. Epileptic Disord. 2012, 14, 371–378. [Google Scholar] [CrossRef]
- Bernath, S. Calcium-independent release of amino acid neurotransmitters: Fact. or artifact? Prog. Neurobiol. 1992, 38, 57–91. [Google Scholar] [CrossRef] [PubMed]
- Niere, F.; Cacheaux, L.P.; Egido-Betancourt, H.X.; Taylor, W.C.; Raab-Graham, K.F. Boosting L-type Ca2+ channel activity in tuberous sclerosis mitigates excess glutamate receptors. BioRxiv 2020. [Google Scholar] [CrossRef]
- Thakur, M.; Rachamalla, M.; Niyogi, S.; Datusalia, A.K.; Flora, S.J.S. Molecular mechanism of arsenic-induced neurotoxicity including neuronal dysfunctions. Int. J. Mol. Sci. 2021, 22, 10077. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xi, S.; Li, X.; Lu, C.; Li, G.; Xu, Y.; Qu, C.; Niu, Y.; Sun, G. Arsenic speciation transported through the placenta from mother mice to their newborn pups. Environ. Res. 2006, 101, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kordas, K.; Lopez, P.; Rosado, J.L.; Cebrian, M.E.; Vargas, G.G.; Ronquillo, D.; Stoltzfus, R.J. Association between arsenic exposure and behavior among first-graders from Torreón, Mexico. Environ. Res. 2011, 111, 670–676. [Google Scholar] [CrossRef]
- Müller, S.M.; Finke, H.; Ebert, F.; Kopp, J.F.; Schumacher, F.; Kleuser, B.; Francesconi, K.A.; Raber, G.; Schwerdtle, T. Arsenic-containing hydrocarbons: Effects on gene expression, epigenetics, and biotransformation in HepG2 cells. Arch. Toxicol. 2018, 92, 1751–1765. [Google Scholar] [CrossRef] [PubMed]
- Bijata, M.; Labus, J.; Guseva, D.; Stawarski, M.; Butzlaff, M.; Dzwonek, J.; Ponimaskin, E. Synaptic remodeling depends on signaling between serotonin receptors and the extracellular matrix. Cell Rep. 2017, 19, 1767–1782. [Google Scholar] [CrossRef]
- Gong, X.W.; Li, J.B.; Lu, Q.C.; Liang, P.J.; Zhang, P.M. Effective connectivity of hippocampal neural network and its alteration in Mg2+-free epilepsy model. PLoS ONE 2014, 9, e92961. [Google Scholar] [CrossRef]
- Zheng, Y.; Tian, C.X.; Dong, L.; Ma, X.X.; Gao, Y.; Xiong, C.; Zhang, K.H.; Li, C.S. Extreme Low Frequency Electromagnetic Field Stimulation Induces Metaplastic-Like Effects on LTP/ LTD. IEEE Access 2019, 7, 152919–152927. [Google Scholar] [CrossRef]
- Xia, P.; Zheng, Y.; Dong, L.; Tian, C. Short-Term Extremely Low-Frequency Electromagnetic Field Inhibits Synaptic Plasticity of Schaffer Collateral-CA1 Synapses in Rat Hippocampus via the Ca2+/Calcineurin Pathway. ACS Chem. Neurosci. 2021, 12, 3550–3557. [Google Scholar] [CrossRef]
- Zheng, Y.; Ma, X.X.; Dong, L.; Ma, W.; Cheng, J.H. Effects of uninterrupted sinusoidal LF-EMF stimulation on LTP induced by different combinations of TBS/HFS at the Schaffer collateral-CA1 of synapses. Brain Res. 2019, 1725, 146487. [Google Scholar] [CrossRef] [PubMed]
- Gloveli, T.; Albrecht, D.; Heinemann, U. Properties of low Mg2+ induced epileptiform activity in rat hippocampal and entorhinal cortex slices during adolescence. Brain Res. Dev. Brain Res. 1995, 87, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Carlen, P.L.; Zhang, L. Examinations of Bilateral Epileptiform Activities in Hippocampal Slices Obtained From Young Mice. Front. Cell Neurosci. 2021, 14, 593840. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Zhao, L.; Tian, L.; Zhao, W.; Xiong, C.; Zheng, Y. AsHC 360 Exposure Influence on Epileptiform Discharges in Hippocampus of Infantile Male Rats In Vitro. Int. J. Mol. Sci. 2023, 24, 16806. https://doi.org/10.3390/ijms242316806
Dong L, Zhao L, Tian L, Zhao W, Xiong C, Zheng Y. AsHC 360 Exposure Influence on Epileptiform Discharges in Hippocampus of Infantile Male Rats In Vitro. International Journal of Molecular Sciences. 2023; 24(23):16806. https://doi.org/10.3390/ijms242316806
Chicago/Turabian StyleDong, Lei, Ling Zhao, Lei Tian, Wenjun Zhao, Chan Xiong, and Yu Zheng. 2023. "AsHC 360 Exposure Influence on Epileptiform Discharges in Hippocampus of Infantile Male Rats In Vitro" International Journal of Molecular Sciences 24, no. 23: 16806. https://doi.org/10.3390/ijms242316806