
Molecular Dynamics and Docking Simulations of Homologous RsmE Methyltransferases Hints at a General Mechanism for Substrate Release upon Uridine Methylation on 16S rRNA


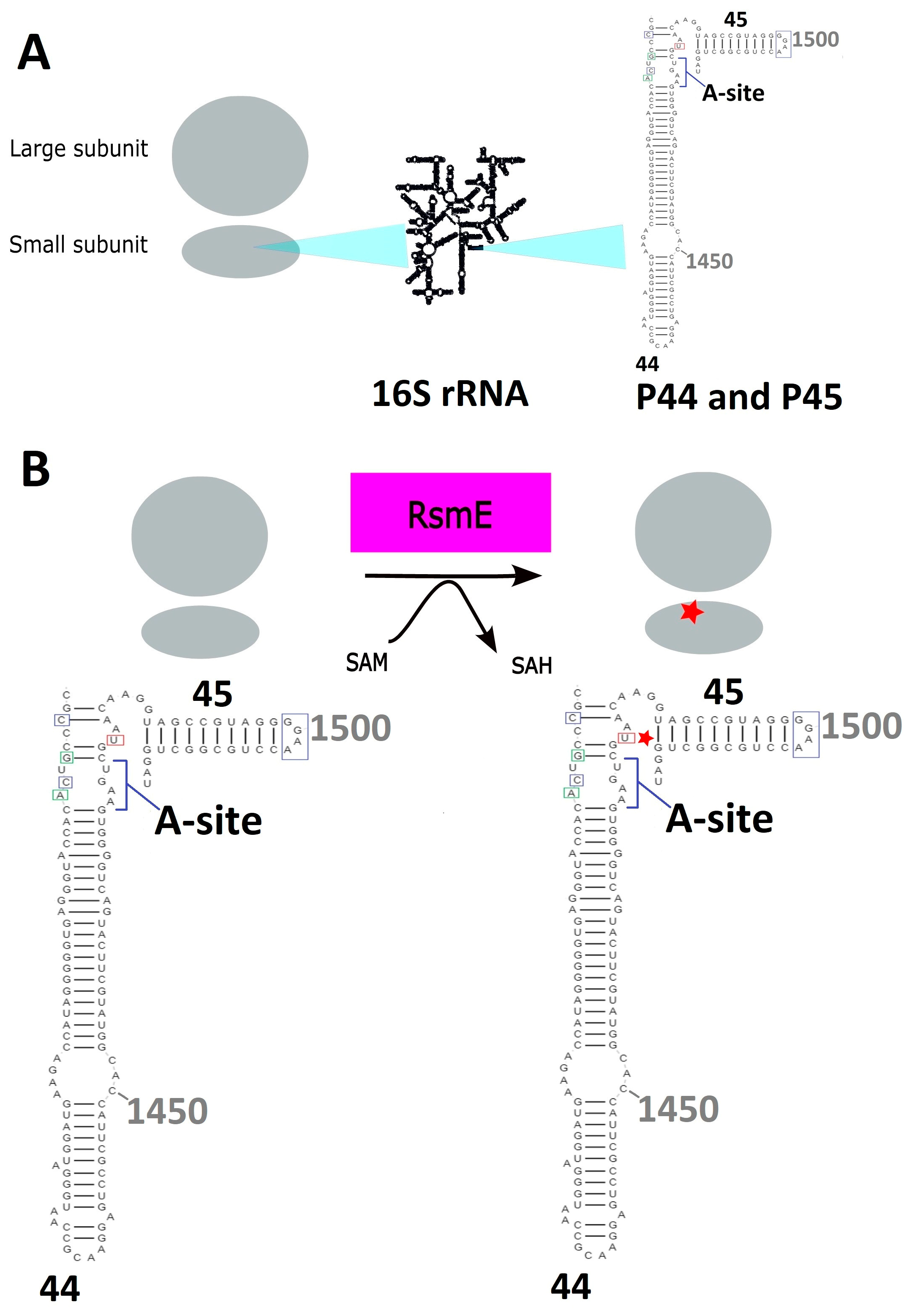{kind=link}
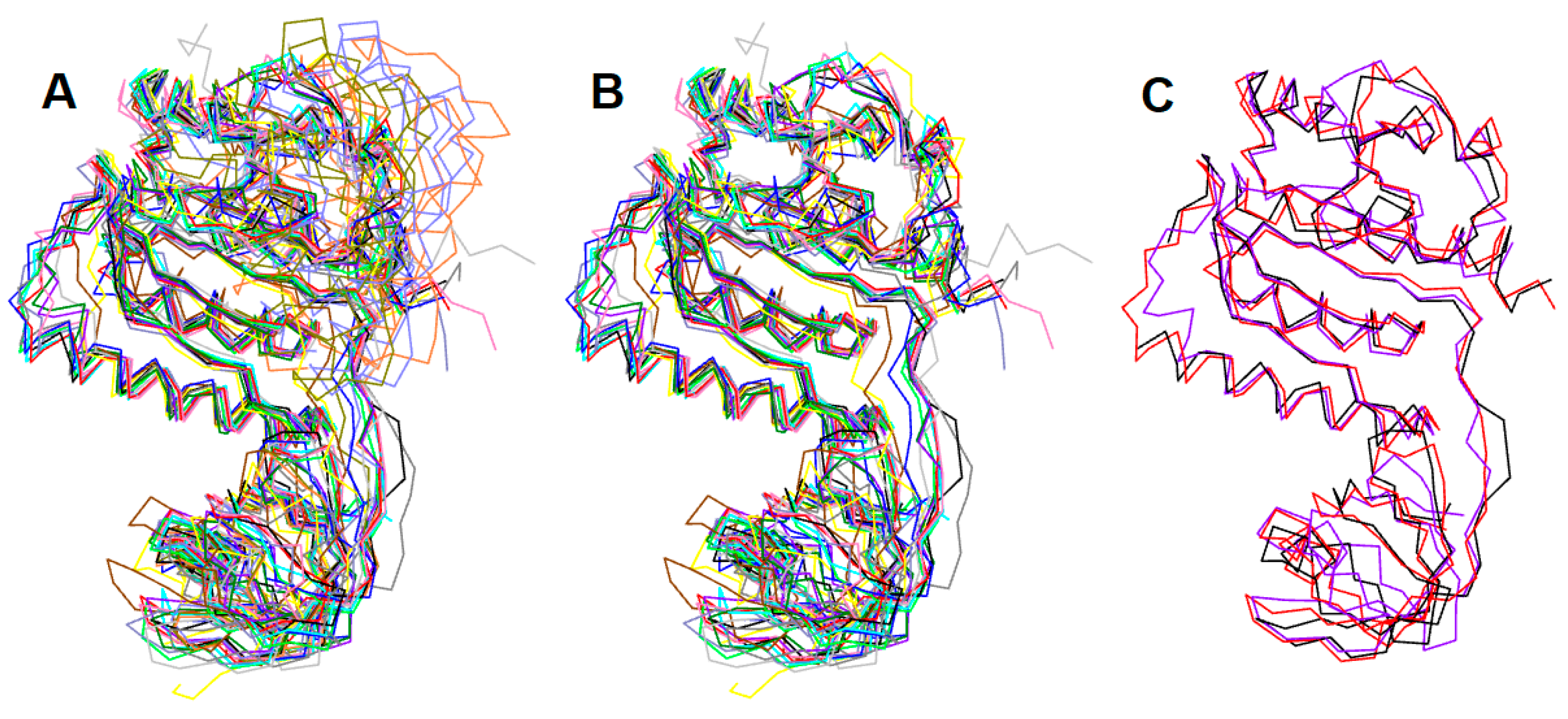{kind=link}
{kind=link}
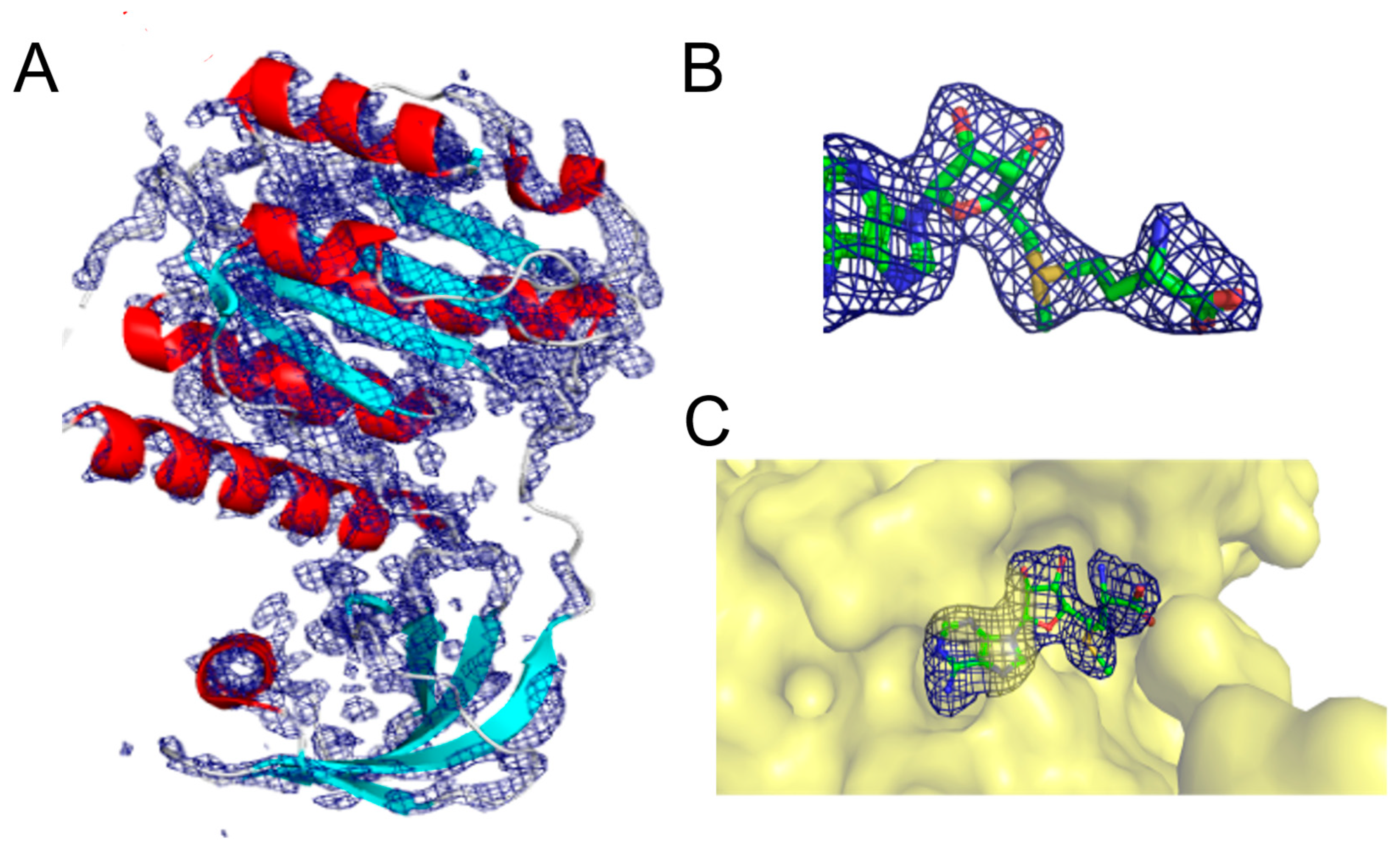{kind=link}
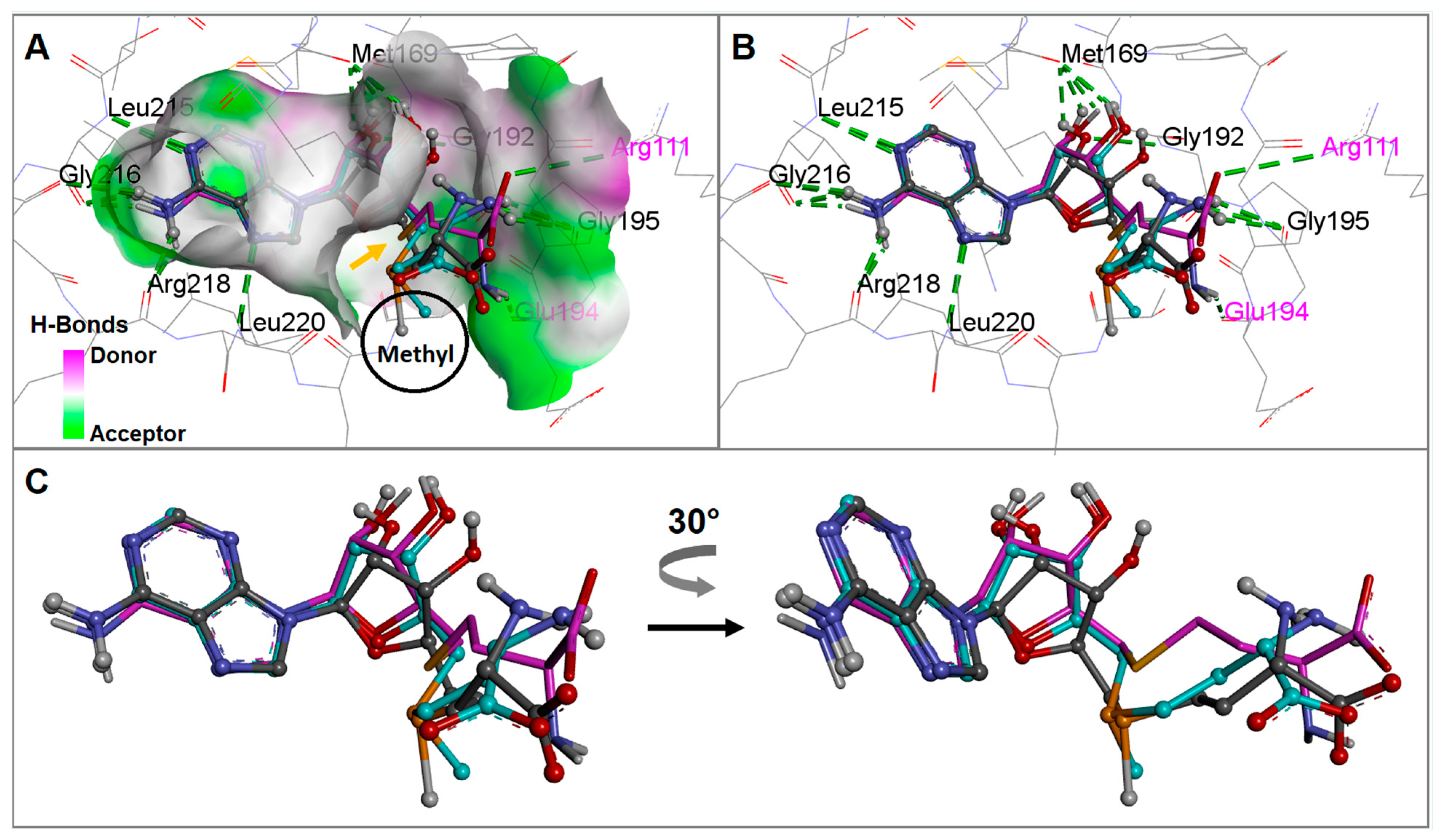{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sequence Alignment and Structural Superposition of the Bacterial RsmEs
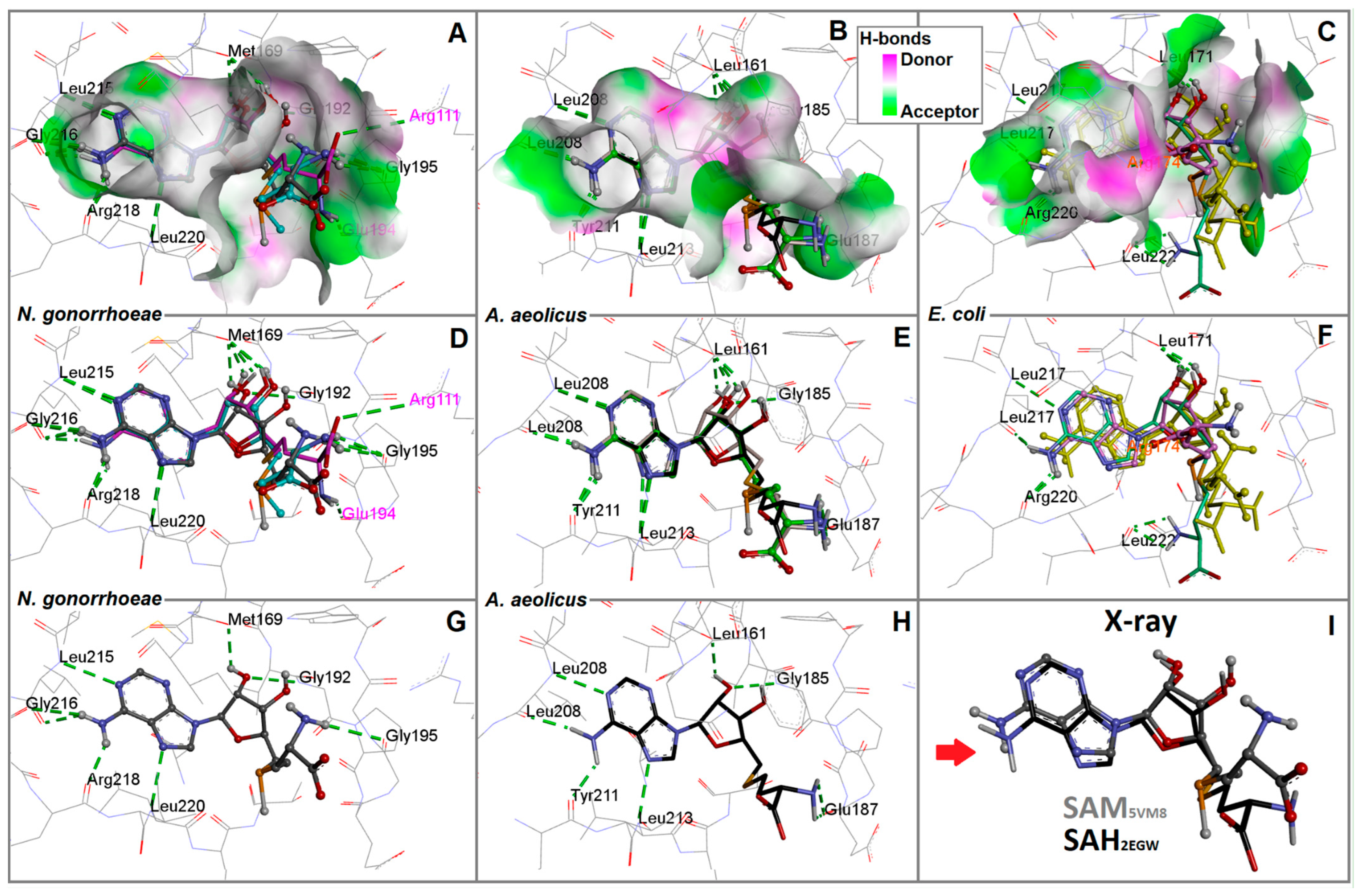
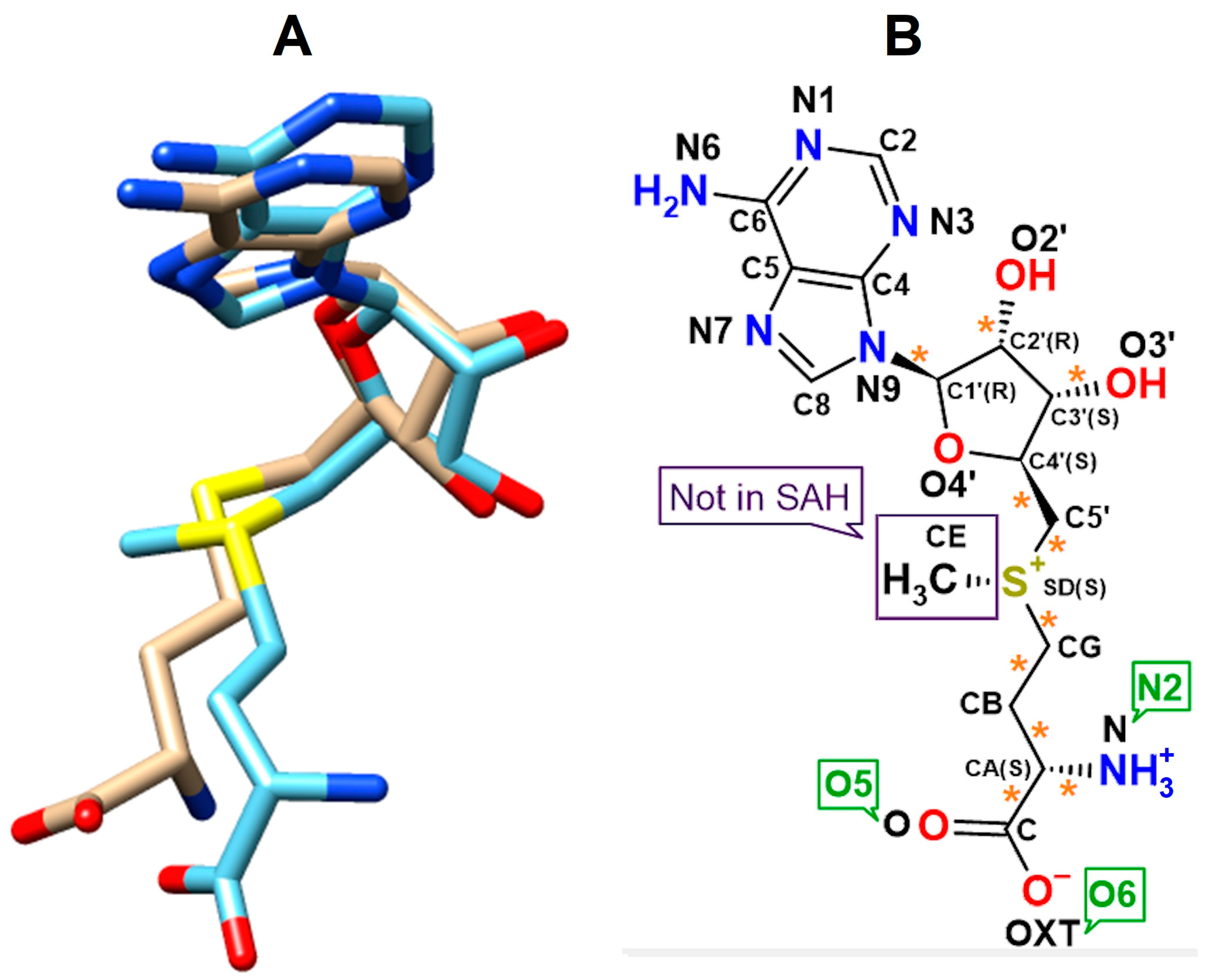
2.2. Interaction Patterns of SAM and SAH in the RsmE Binding Site of N. gonorrhoeae from the Molecular Dockings and the Crystal Structure
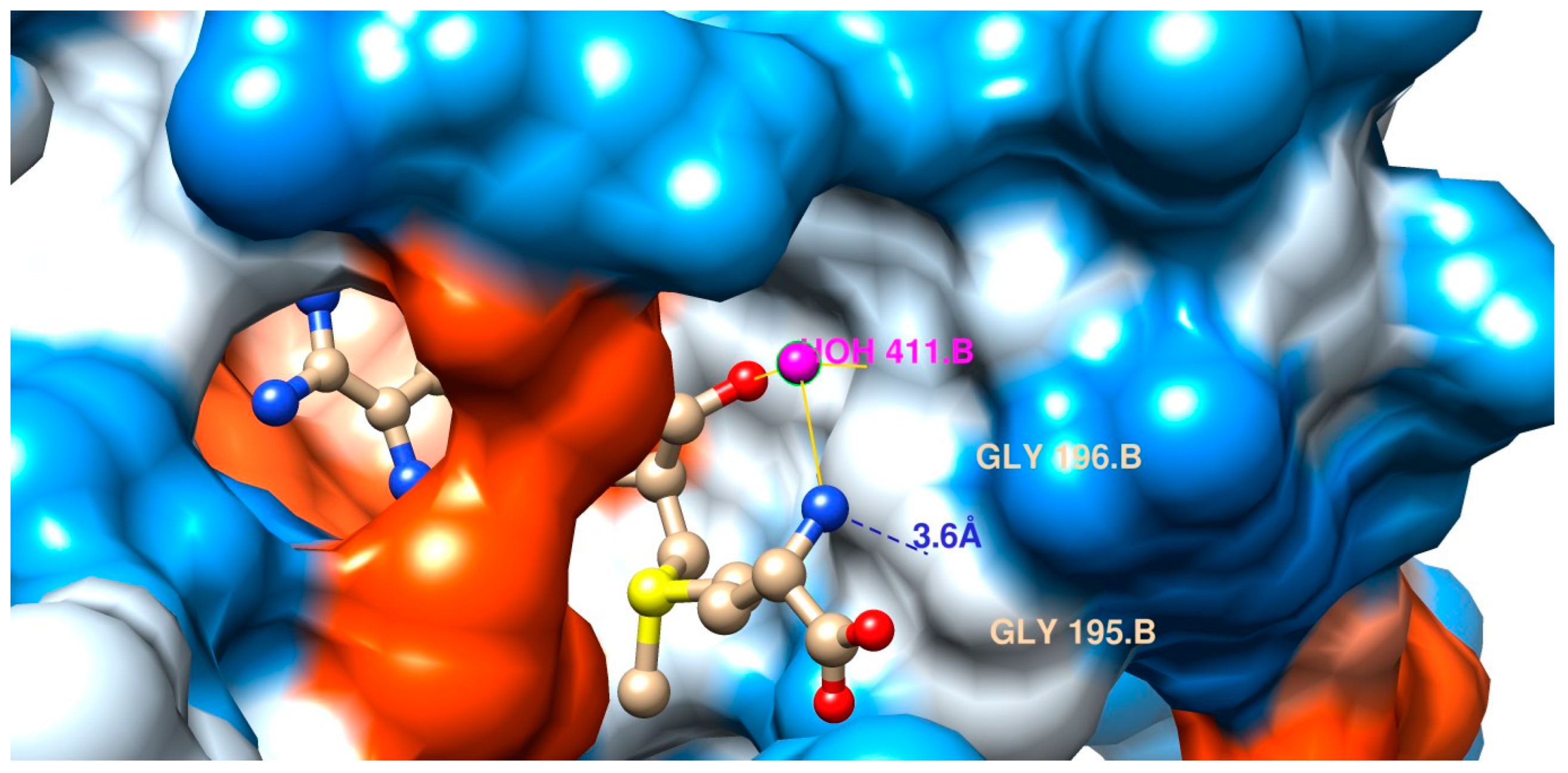
2.3. The Bacterial RsmE Binding Site Is a Crevice with a Network of Hydrogen Bonds and Hydrophobic Interactions
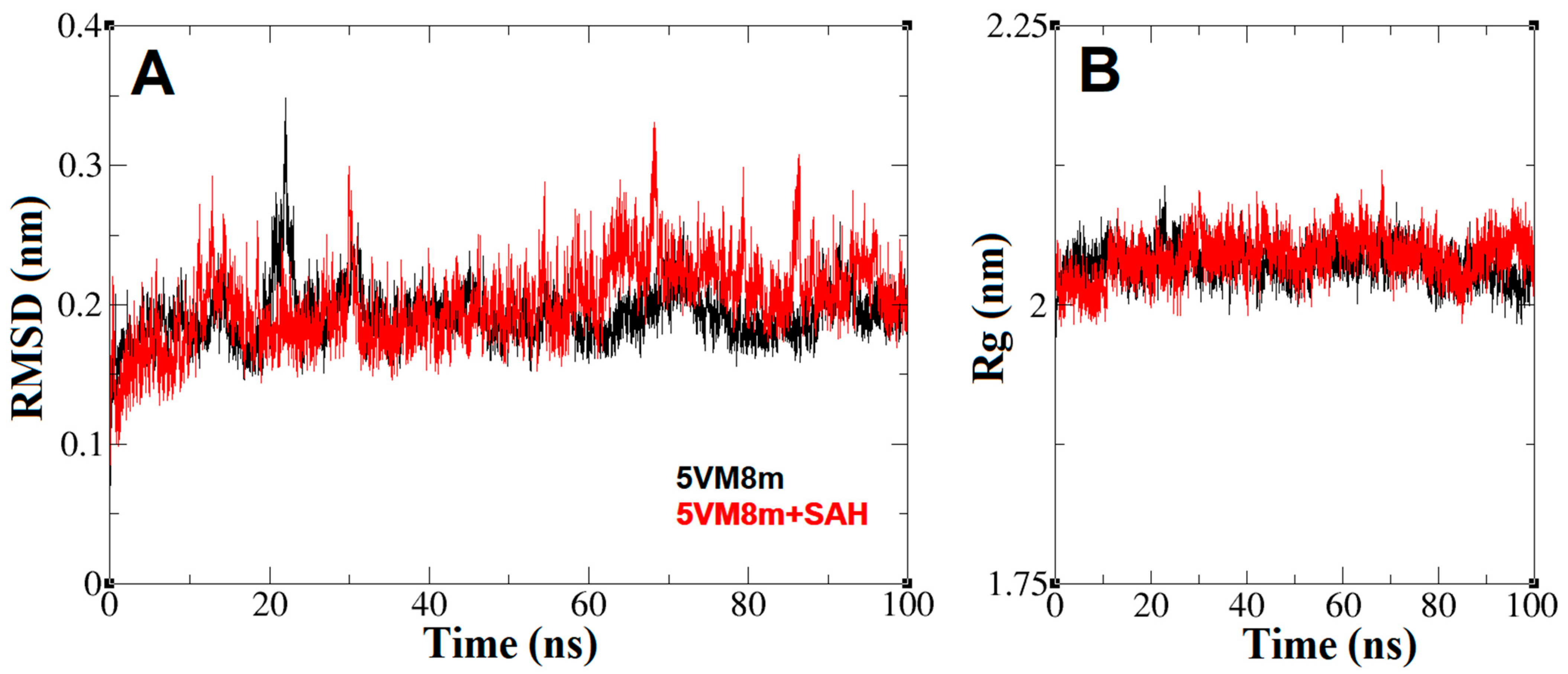
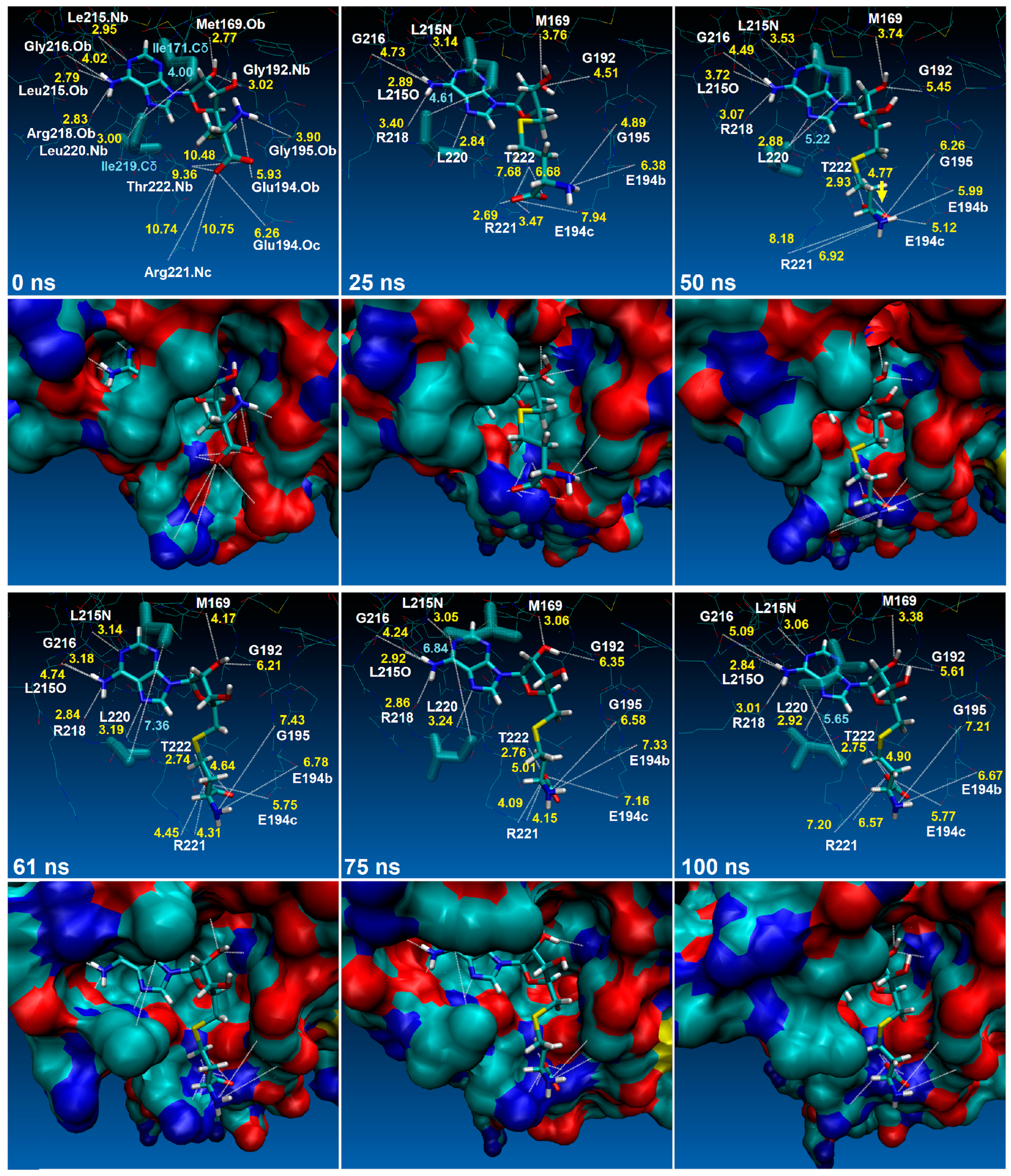
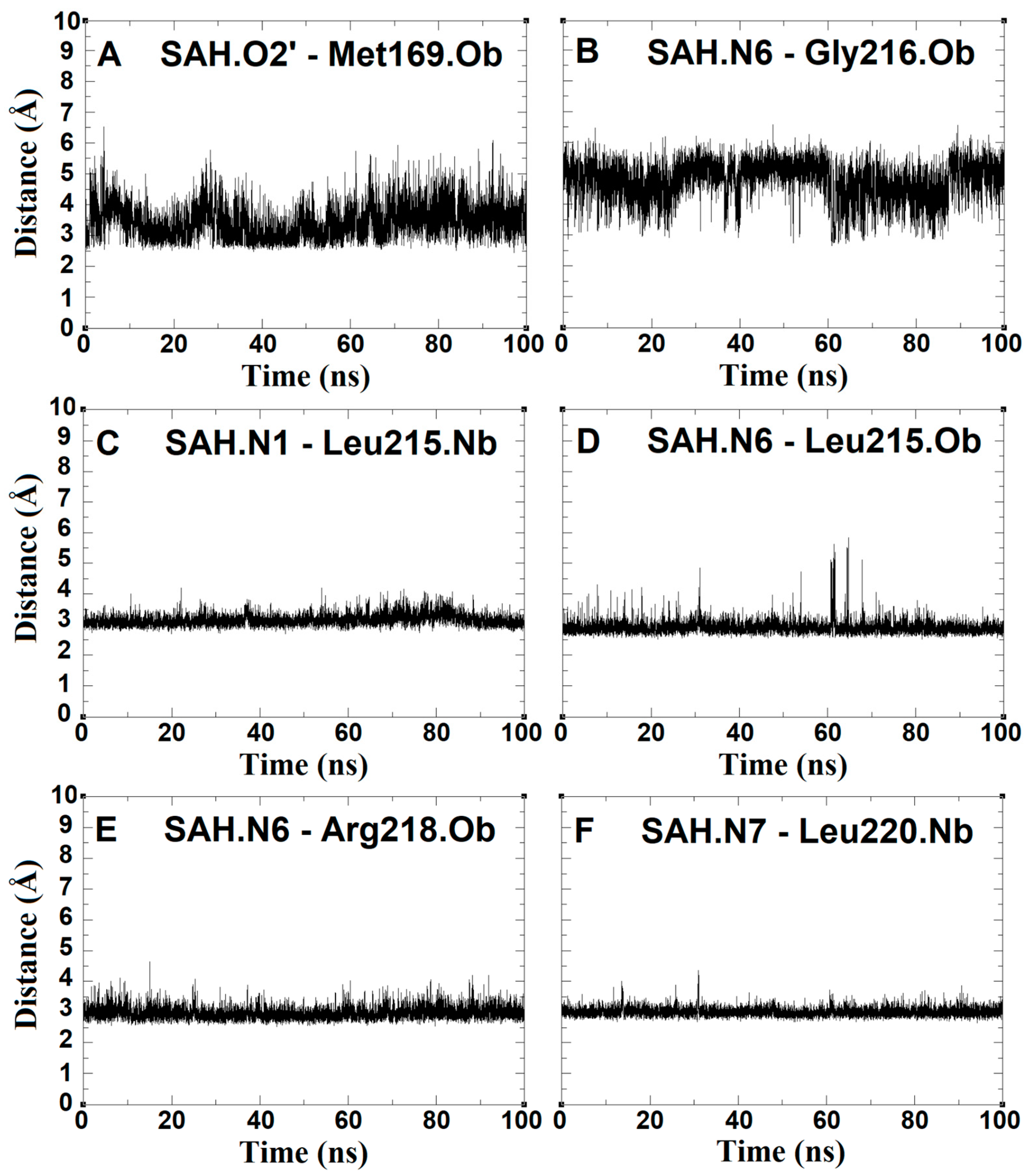
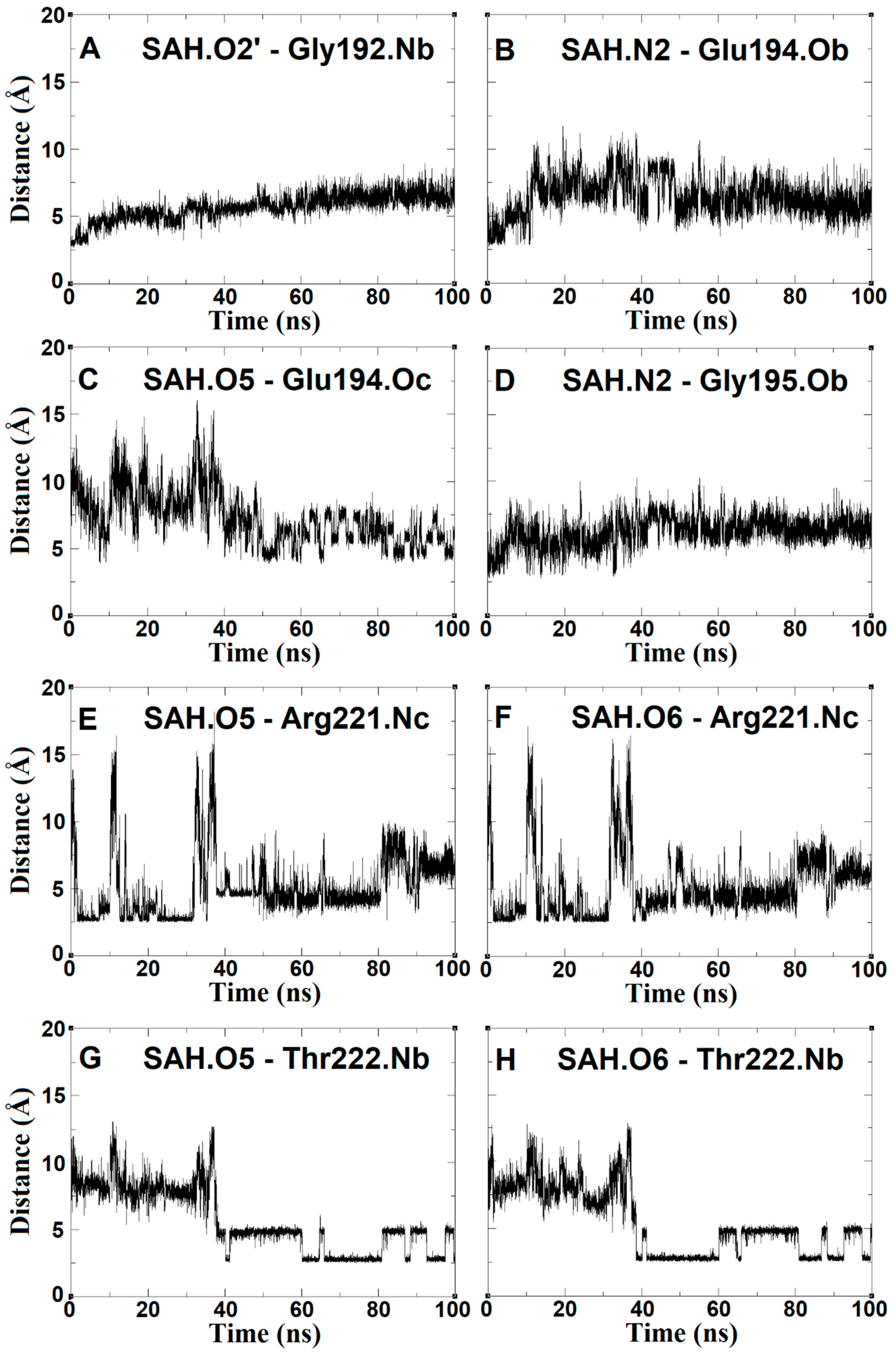
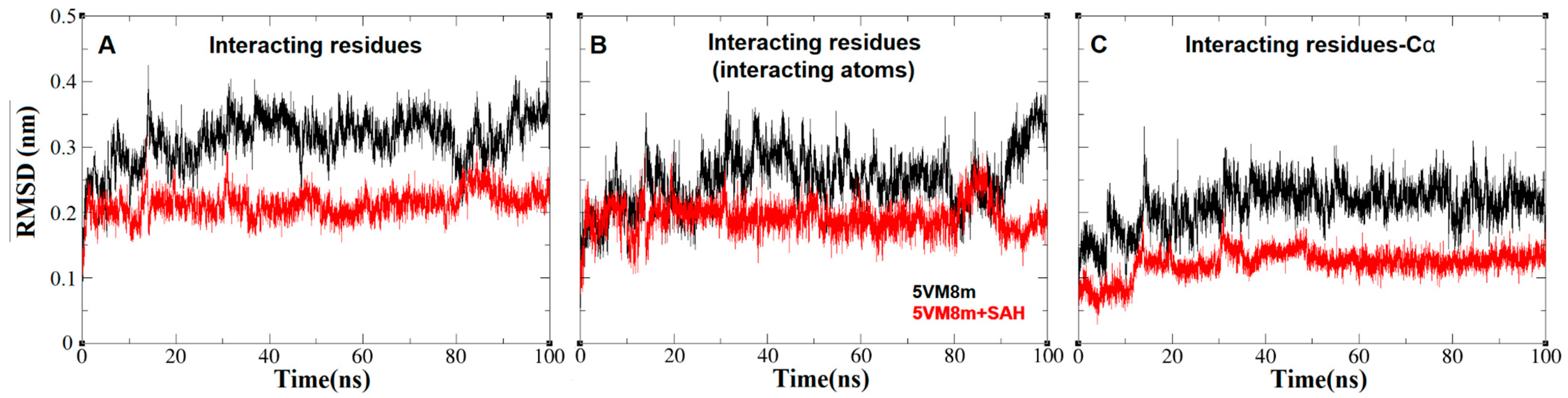
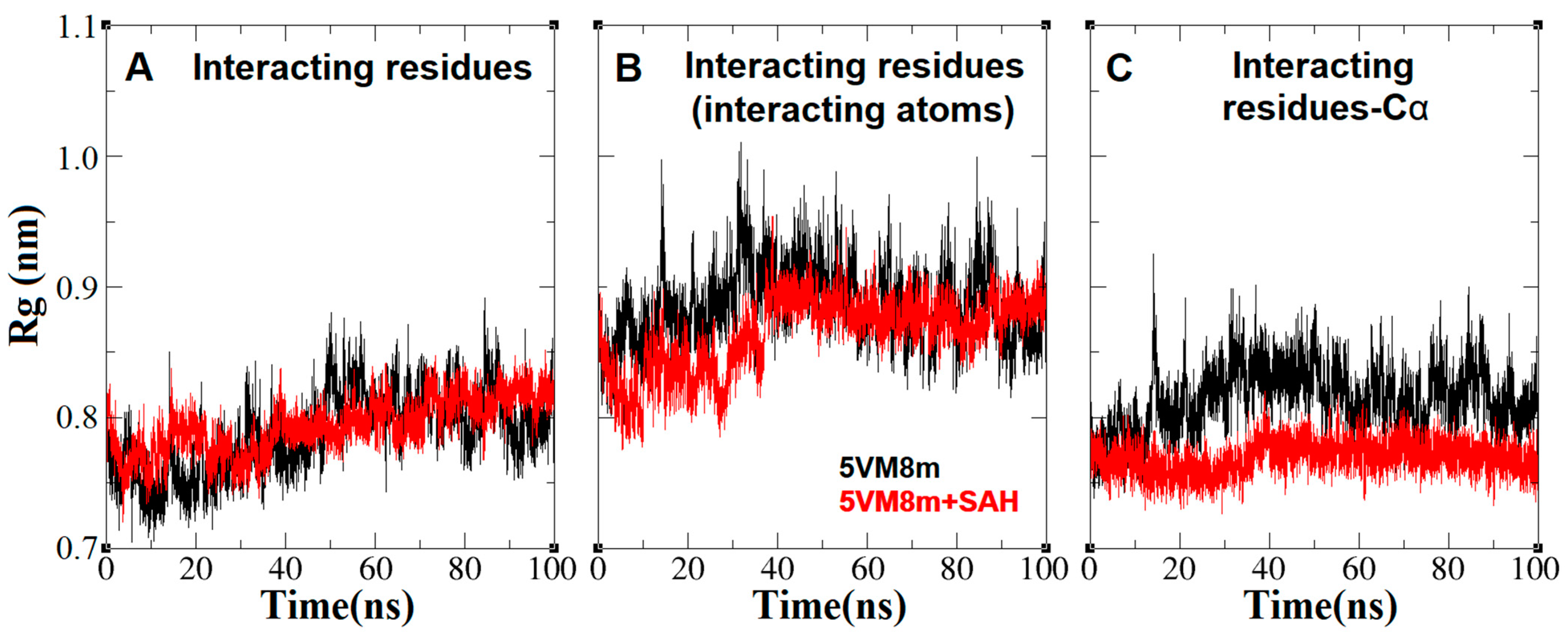
2.4. The Dynamics inside RsmE during SAH Release
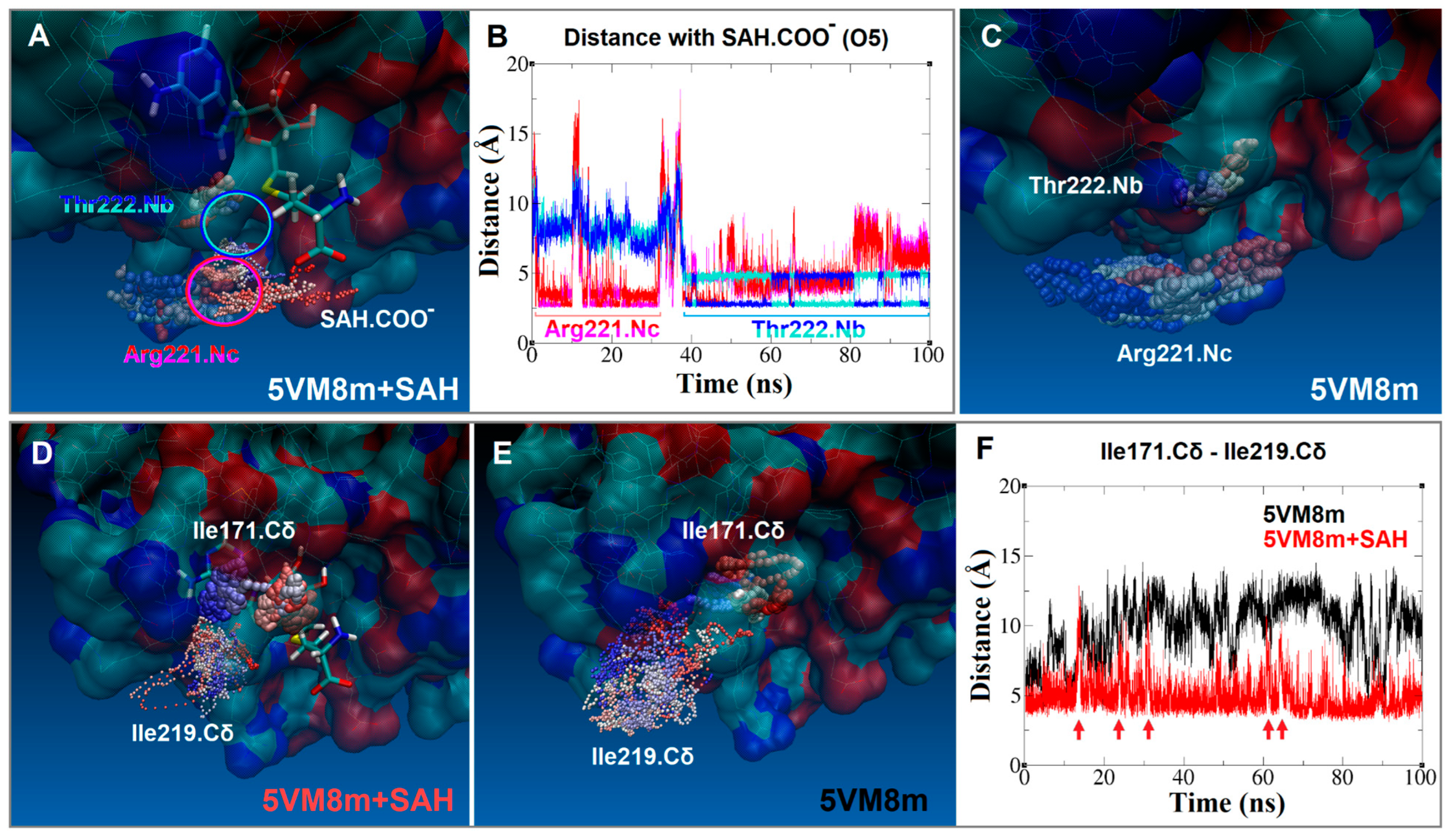
2.5. SAH Exchange between Arg221 and Thr222 Is Based on Swinging Side Chains
2.6. An Isoleucine Gate Is Active in Concert with Arg/Thr Contacts to Release SAH
3. Discussion
3.1. Literature-Based Evidence and Molecular Dynamics Results
3.2. Structural Insight and Bacterial Resistance
4. Materials and Methods
4.1. RsmE Multiple Sequence Alignment and Molecular Structure Superposition
4.2. Molecular Design for Computational Simulations
4.3. Molecular Docking of SAM and SAH at the RsmE Binding Site of N. gonorrhoeae, A. aeolicus and E. coli
4.4. Molecular Dynamics of Monomeric N. gonorrhoeae RsmE Alone and with SAH
4.5. Model Limitations and Study Design Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basturea, G.N.; Rudd, K.E.; Deutscher, M.P. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 2006, 12, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wan, H.; Gao, Z.Q.; Wei, Y.; Wang, W.J.; Liu, G.F.; Shtykova, E.V.; Xu, J.H.; Dong, Y.H. Insights into the Catalytic Mechanism of 16S rRNA Methyltransferase RsmE (m3U1498) from Crystal and Solution Structures. J. Mol. Biol. 2012, 423, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sanchez, M.I.G.; Cipullo, M.; Gopalakrishna, S.; Khawaja, A.; Rorbach, J. Methylation of Ribosomal RNA: A Mitochondrial Perspective. Front. Genet. 2020, 11, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Zarubica, T.; Baker, M.R.; Wright, H.T.; Rife, J.P. The aminoglycoside resistance methyltransferases from the ArmA/Rmt family operate late in the 30S ribosomal biogenesis pathway. RNA 2011, 17, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Duss, O.; Michel, E.; Diarra dit Konté, N.; Schubert, M.; Allain, F.H. Molecular bases for the wide range of affinity found in Csr/Rsm protein-RNA recognition. Nucleic Acids Res. 2014, 8, 5332–5346. [Google Scholar] [CrossRef] [PubMed]
- Golovina, A.Y.; Dzama, M.M.; Osterman, I.A.; Sergiev, P.V.; Serebryakova, M.V.; Bogdanov, A.A.; Dontsova, O.A. The last rRNA methyltransferase of E. coli revealed: The yhiR gene encodes adenine-N6 methyltransferase specific for modification of A2030 0f 23S ribosomal RNA. RNA 2012, 18, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Basturea, G.N.; Deutscher, M.P. Substrate specificity and properties of the Escherichia coli 16S rRNA methyltransferase, RsmE. RNA 2007, 13, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Arakawa, Y. 16S Ribosomal RNA Methylation: Emerging Resistance Mechanism against Aminoglycosides. Clin. Infect. Dis. 2007, 45, 88–94. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Pfeiffer, M.; Ribar, A.; Nidetzky, B. A selective and atom-economic rearrangement of uridine by cascade biocatalysis for production of pseudouridine. Nat. Commun. 2023, 14, 2261–2274. [Google Scholar] [CrossRef]
- Dimitrova, D.G.; Teysset, L.; Carré, C. RNA 2-O-Methylation (Nm) Modification in Human Diseases. Genes 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, S.; Taneja, B. The structure of Rv2372c identifies an RsmE-like methyltransferase from Mycobaterium tuberculosis. Acta Cryst. 2014, D70, 821–832. [Google Scholar]
- Wasif Baig, M.; Pederzoli, M.; Kývala, M.; Cwiklik, L.; Pittner, J. Theoretical investigation of the effect of alkylation and bromination on intersystem crossing in BODIPY-based photosensitizers. J. Phys. Chem. B 2021, 125, 11617–11627. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.V.; Bogdanov, A.A.; Sergiev, P.V. What do we know about ribosomal RNA methylation in Escherichia coli? Biochimie 2015, 117, 110–118. [Google Scholar] [CrossRef]
- Mokmak, W.; Tongsima, S.; Jenwitheesuk, E. Molecular dynamics simulation of a human thiopurine S-methyltransferase complexed with 6-mercaptopurine model. Bioinformation 2009, 4, 59–62. [Google Scholar] [CrossRef]
- Sergiev, P.; Aleksashin, N.; Chugunova, A.; Polikanov, Y.S.; Dontsova, O.A. Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 2018, 14, 226–235. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Clemons, W.M., Jr.; Carter, A.P.; Winverly, B.T.; Ramakrishnan, V. Crystal Structure of the 30 S ribosomal subunit from Thermus thermophilus: Structure of the proteins and their interaction with 16 S RNA. J. Mol. Biol. 2002, 316, 725–768. [Google Scholar] [CrossRef]
- Pinotsis, N.; Waksman, G. Crystal structure of the Legionella pneumophilia Lpg2936 in complex with the cofactor S-adenosyl-L-methionine reveals novel insights into the mechanism of RsmE family methyltransferases. Protein Sci. 2017, 26, 2381–2391. [Google Scholar] [CrossRef]
- Burroughs, A.M.; Allen, K.N.; Dunaway-Mariano, D.; Aravind, L. Evolutionary genomics of the HAD superfamily: Understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 2006, 361, 1003–1034. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef]
- Forouhar, F.; Shen, J.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Tong, L. Functional assignment based on structural analysis: Crystal structure of the yggJ protein (HI0303) of Haemophilus influenzae reveals an RNA methyltransferase with a deep trefoil knot. Proteins 2003, 53, 329–332. [Google Scholar] [CrossRef]
- Badger, J.; Sauder, J.M.; Adams, J.M.; Antonysamy, S.; Bain, K.; Bergseid, M.G.; Buchanan, S.G.; Buchanan, M.D.; Batiyenko, Y.; Christopher, J.A.; et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 2005, 60, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, I.; Scior, T. Induced fit for cytochrome P450 3A4 based on molecular dynamics. ADMET DMPK 2019, 7, 252–266. [Google Scholar] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Dueñas-González, A.; Lyko, F.; Medina-Franco, J.L. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. Chem. Med. Chem. 2009, 4, 792–799. [Google Scholar] [CrossRef]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef]
- Gsponer, J.; Caflisch, A. Molecular dynamics simulations of protein folding from the transition state. Comput. Biol. Chem. 2002, 99, 6719–6724. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Saloni; Misra, S.; Yadav, L.; Teli, M.; Sharma, P.; Chaudhary, S.; Kumar, N.; Choi, E.H.; et al. Molecular Insights into the Interaction of RONS and Thieno[3,2-c]pyran Analogs with SIRT6/COX-2: A Molecular Dynamics Study. Sci. Rep. 2018, 8, 4777. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D1. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 2010, 38, w695–w699. [Google Scholar] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [PubMed]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: An versatile platform for cheminformatics and drug design projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar]
- NIH—National Library of Medicine—Drugbank. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/S-Adenosyl-L-methionine#section=IUPAC-Name (accessed on 25 June 2023).
- NIH—National Library of Medicine—Drugbank. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/S-Adenosyl-L-homocysteine#section=IUPAC-Name (accessed on 25 June 2023).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [PubMed]
- Scior, T.; Verhoff, M.; Gutierrez-Aztatzi, I.; Ammon, H.P.; Laufer, S.; Werz, O. Interference of boswellic acids with the ligand binding domain of the glucocorticoid receptor. J. Chem. Inf. Model 2014, 54, 978–986. [Google Scholar]
- Scior, T.; Lozano-Aponte, J.; Ajmani, S.; Hernández-Montero, E.; Chávez-Silva, F.; Hernández-Núñez, E.; Moo-Puc, R.; Fraguela-Collar, A.; Navarrete-Vázquez, G. Antiprotozoal Nitazoxanide Derivatives: Synthesis, Bioassays and QSAR Study Combined with Docking for Mechanistic Insight. Curr. Comput. Aided Drug Des. 2015, 11, 21–31. [Google Scholar]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer, version 20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model 2012, 52, 3144–3154. [Google Scholar]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52, 3155–3168. [Google Scholar]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Soteras Gutiérrez, I.; Lin, F.Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L., 3rd; MacKerell, A.D., Jr. Parametrization of Halogen Bonds in the CHARMM General Force Field: Improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open-source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software-X 2015, 1, 19–25. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Turner, P.J. XMGRACE; Version 5.1.19; Center for Coastal and Land-Margin Research, Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Murayama, K.; Shirouzu, M.; Yokoyama, S. Crystal Sstructure of TT1573 from Thermus thermophilus. 2004. Available online: https://doi.org/10.2210/pdb1V6Z/pdb (accessed on 6 September 2023).
- Joint Center for Structural Genomics. Crystal Structure of Hypothetical Protein (TM1380) from Thermotoga maritima at 2.12 A Resolution. 2005. Available online: https://doi.org/10.2210/pdb1Z85/pdb (accessed on 6 September 2023).
- Murayama, K.; Shirouzu, M.; Yokoyama, S. Crystal Structure of Methyltransferase with Ligand (SAH). 2005. Available online: https://doi.org/10.2210/pdb2CX8/pdb (accessed on 6 September 2023).
- Hikida, Y.; Kuratani, M.; Bessho, Y.; Sekine, S.I.; Yokoyama, S. Structure of an archaeal homologue of the bacterial Fmu/RsmB/RrmB rRNA cytosine 5-methyltransferase. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 1301–1307. [Google Scholar] [CrossRef]
- Murayama, K.; Terada, T.; Kuramitsu, S.; Shirouzu, M.; Yokoyama, S.; RIKEN Structural Genomics/Proteomics Initiative (RSGI). Crystal Structure of the TTHA0657-SAM Complex. 2007. Available online: https://doi.org/10.2210/pdb2Z0Y/pdb (accessed on 6 September 2023).
- Ramagopal, U.A.; Toro, R.; Burley, S.K.; Almo, S.C. Crystal Structure of Probable rRNA-Methyltransferase from Porphy-Romonas Gingivalis. 2010. Available online: https://doi.org/10.2210/pdb3KW2/pdb (accessed on 6 September 2023).
- Eswaramoorthy, S.; Almo, S.C.; Swaminathan, S. Crystal Structure of 16S Ribosomal RNA Methyltransferase RsmE. 2013. Available online: https://doi.org/10.2210/pdb4J3C/pdb (accessed on 6 September 2023).
- Available online: https://go.drugbank.com/drugs/DB00118 (accessed on 6 September 2023).
- Available online: https://go.drugbank.com/drugs/DB01752 (accessed on 6 September 2023).
- Husain, N.; Obranic, S.; Koscinski, L.; Seetharaman, J.; Babic, F.; Bujnicki, J.M.; Maravic-Vlahovicek, G.; Sivaraman, J. Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res. 2011, 39, 1903–1918. [Google Scholar]
- Smith, R.D.; Clark, J.J.; Ahmed, A.; Orban, Z.J.; Dunbar, J.B., Jr.; Carlson, H.A. Updates to Binding MOAD (Mother of All Data-bases): Polypharmacology Tools and Their Utility in Drug Repurposing. J. Mol. Biol. 2019, 431, 2423–2433. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.Y.; Wang, S. The PDBbind database: Mmethodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef]
- Wermuth, C. Chapter 12. Lead-Likeness and Drug-Likeness. In The Practice of Medicinal Chemistry, 3rd ed.; Academic Press, Prestwick Chemical Inc.: Illkirch, France, 2008; p. 250. [Google Scholar]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Cid, A.; Lozano-Aponte, J.; Scior, T. Molecular Dynamics and Docking Simulations of Homologous RsmE Methyltransferases Hints at a General Mechanism for Substrate Release upon Uridine Methylation on 16S rRNA. Int. J. Mol. Sci. 2023, 24, 16722. https://doi.org/10.3390/ijms242316722
Hernández-Cid A, Lozano-Aponte J, Scior T. Molecular Dynamics and Docking Simulations of Homologous RsmE Methyltransferases Hints at a General Mechanism for Substrate Release upon Uridine Methylation on 16S rRNA. International Journal of Molecular Sciences. 2023; 24(23):16722. https://doi.org/10.3390/ijms242316722
Chicago/Turabian StyleHernández-Cid, Aaron, Jorge Lozano-Aponte, and Thomas Scior. 2023. "Molecular Dynamics and Docking Simulations of Homologous RsmE Methyltransferases Hints at a General Mechanism for Substrate Release upon Uridine Methylation on 16S rRNA" International Journal of Molecular Sciences 24, no. 23: 16722. https://doi.org/10.3390/ijms242316722