
Time-Course Transcriptome Analysis of Aquilegia vulgaris Root Reveals the Cell Wall’s Roles in Salinity Tolerance
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Results of Transcriptome Sequencing and Assessment of Quality
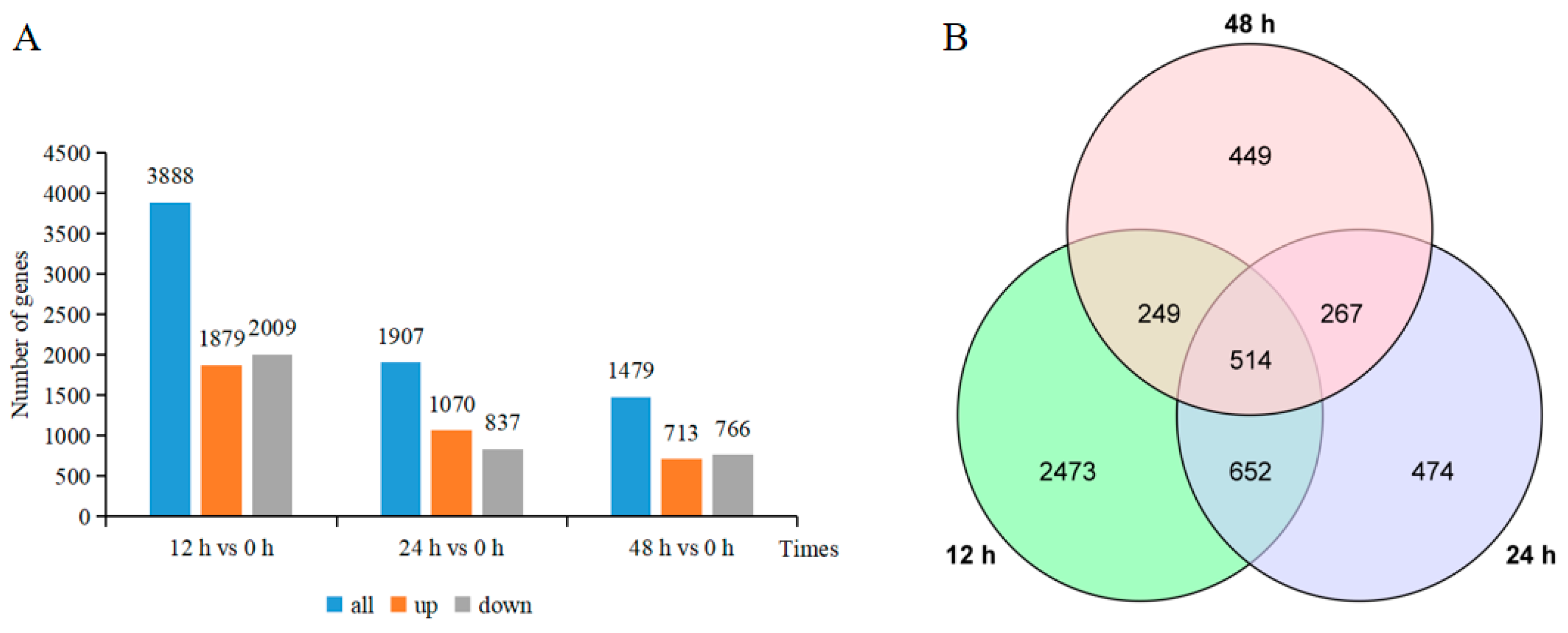
2.2. Investigation of the Differentially Expressed Genes (DEGs)
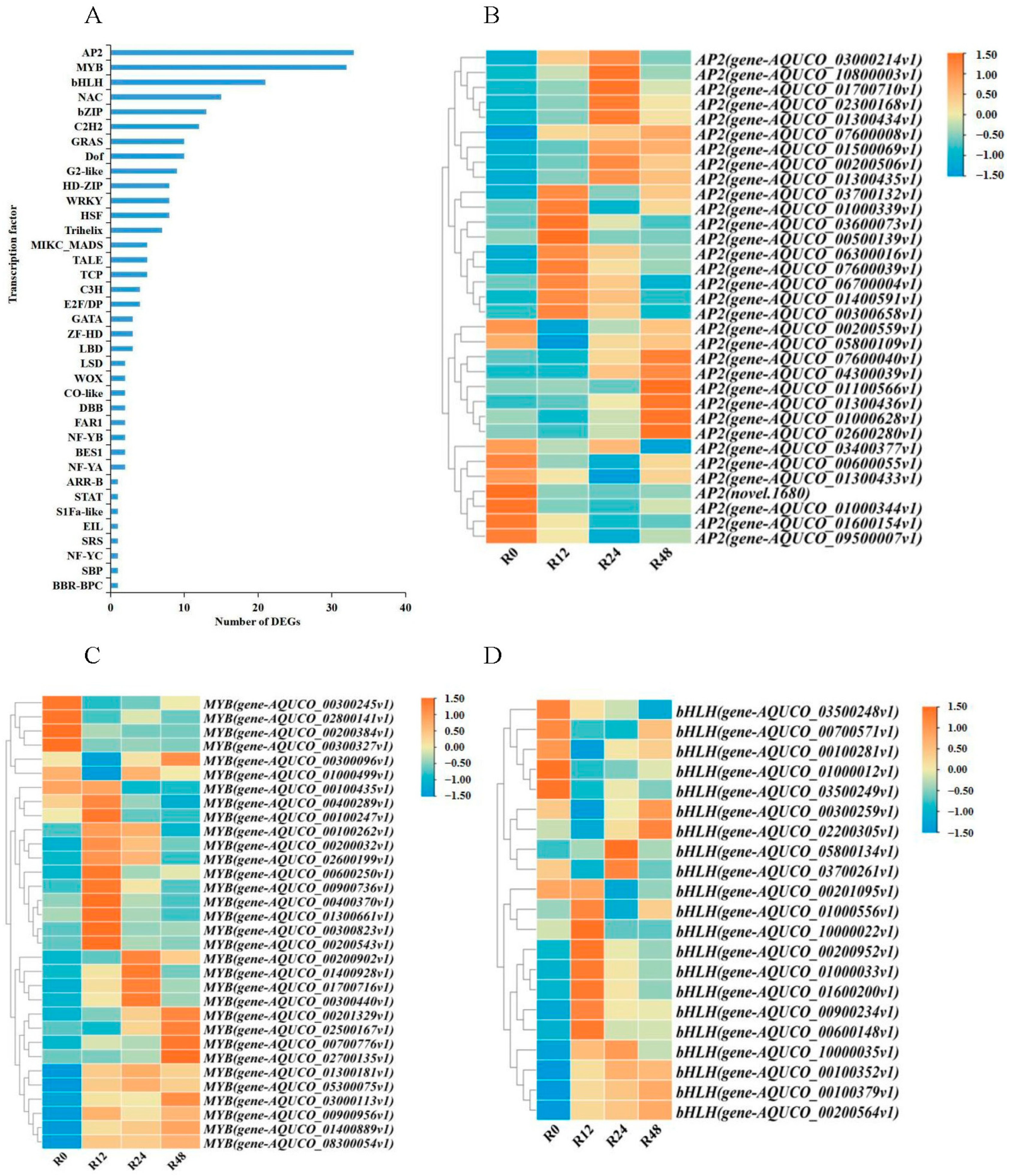
2.3. Analysis of Expression and Enrichment of TFs among DEGs
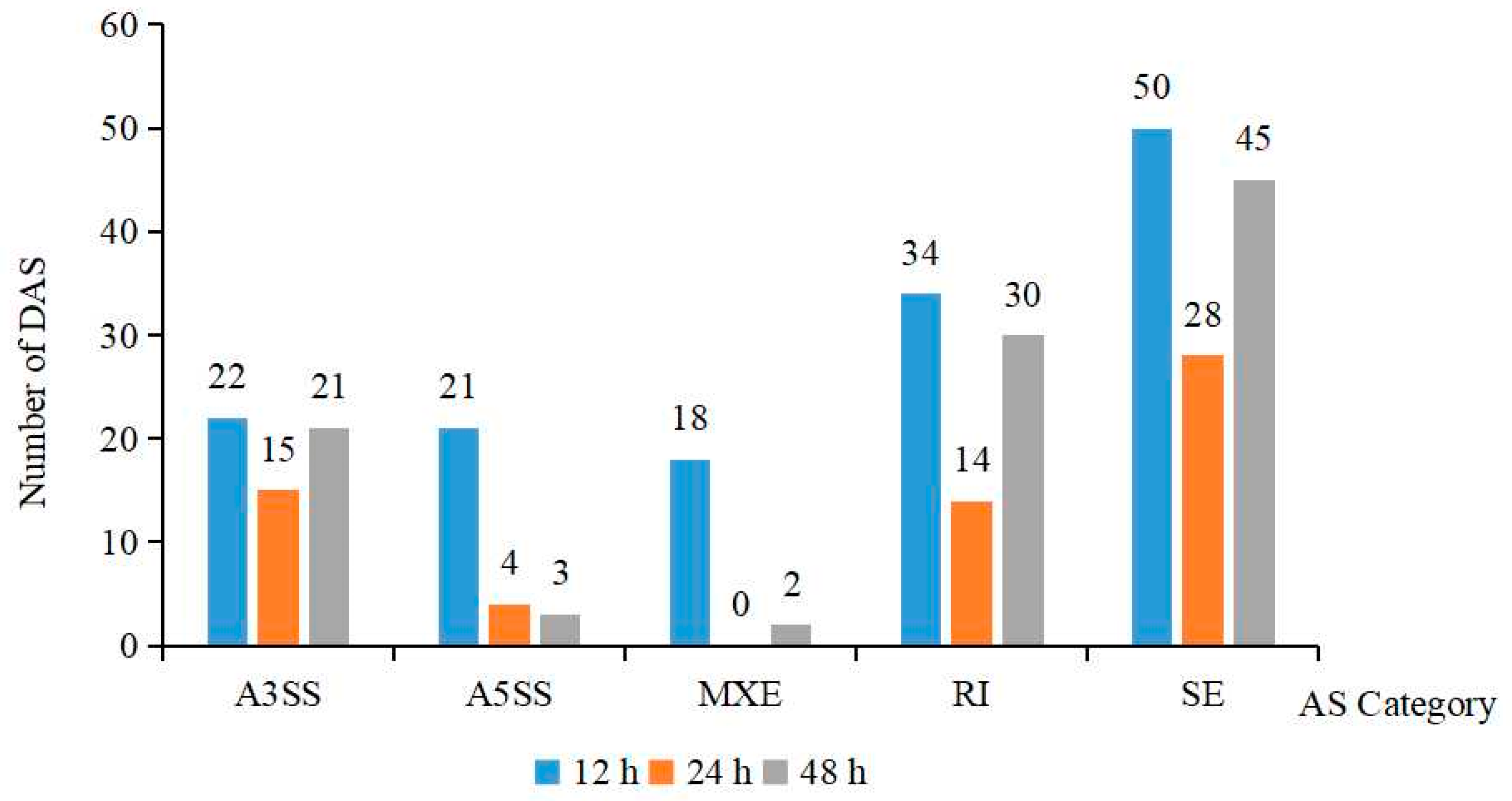
2.4. Analysis of Differences in Alternative Splicing (DAS)
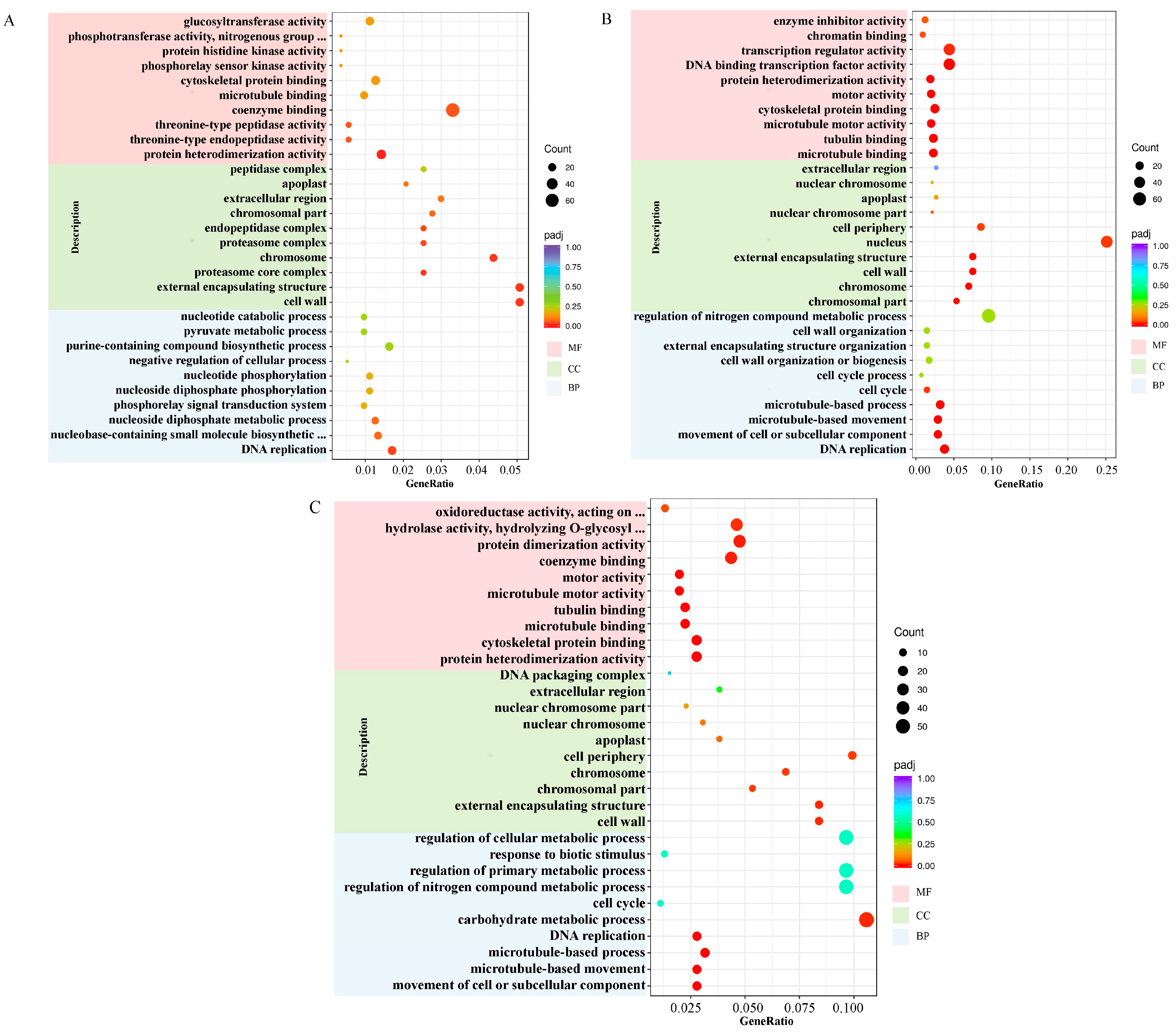
2.5. Enrichment Analysis of Gene Ontology (GO) Terms for DEGs
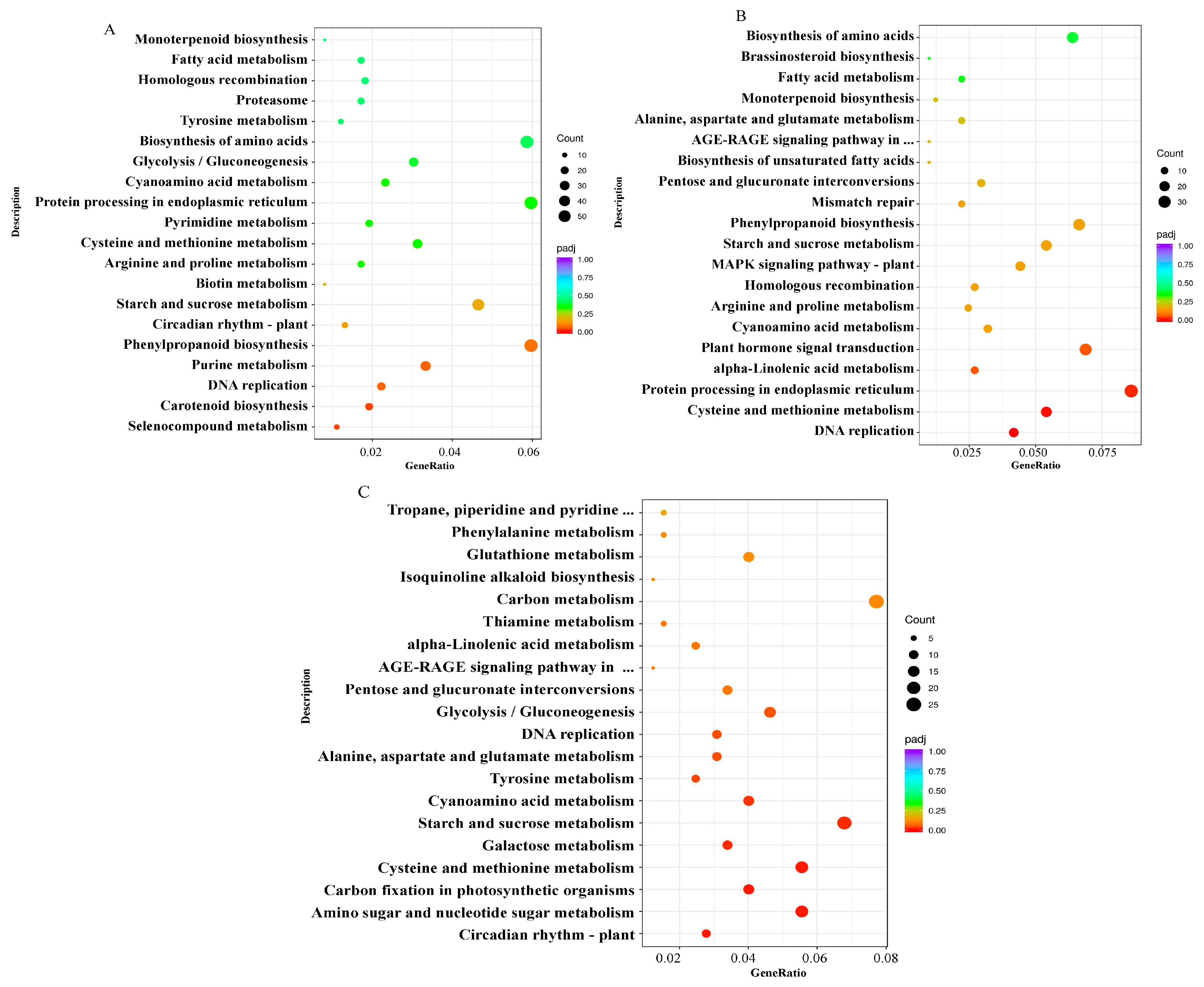
2.6. Enrichment Analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways for DEGs
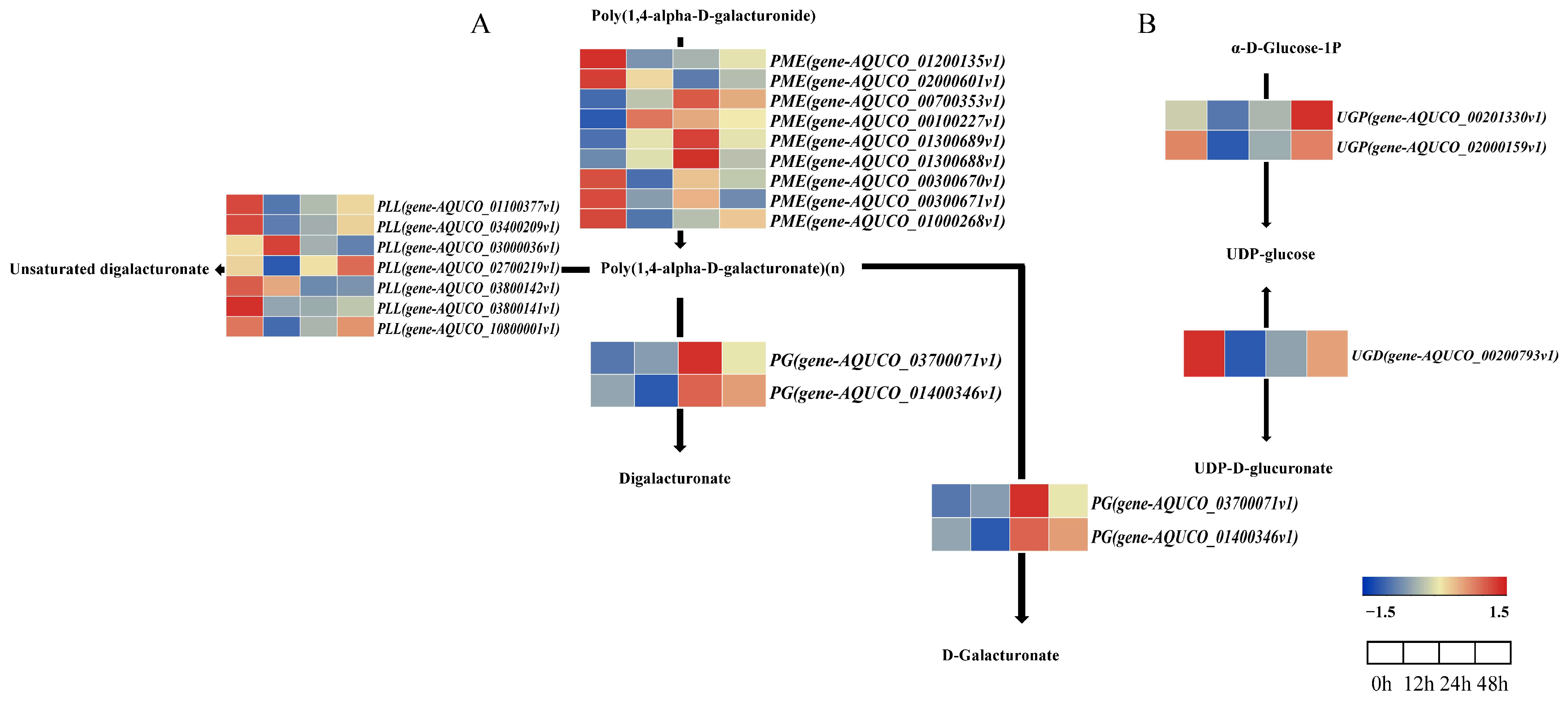
2.7. Analysis of the Pentose and Glucuronate Interconversion Pathway
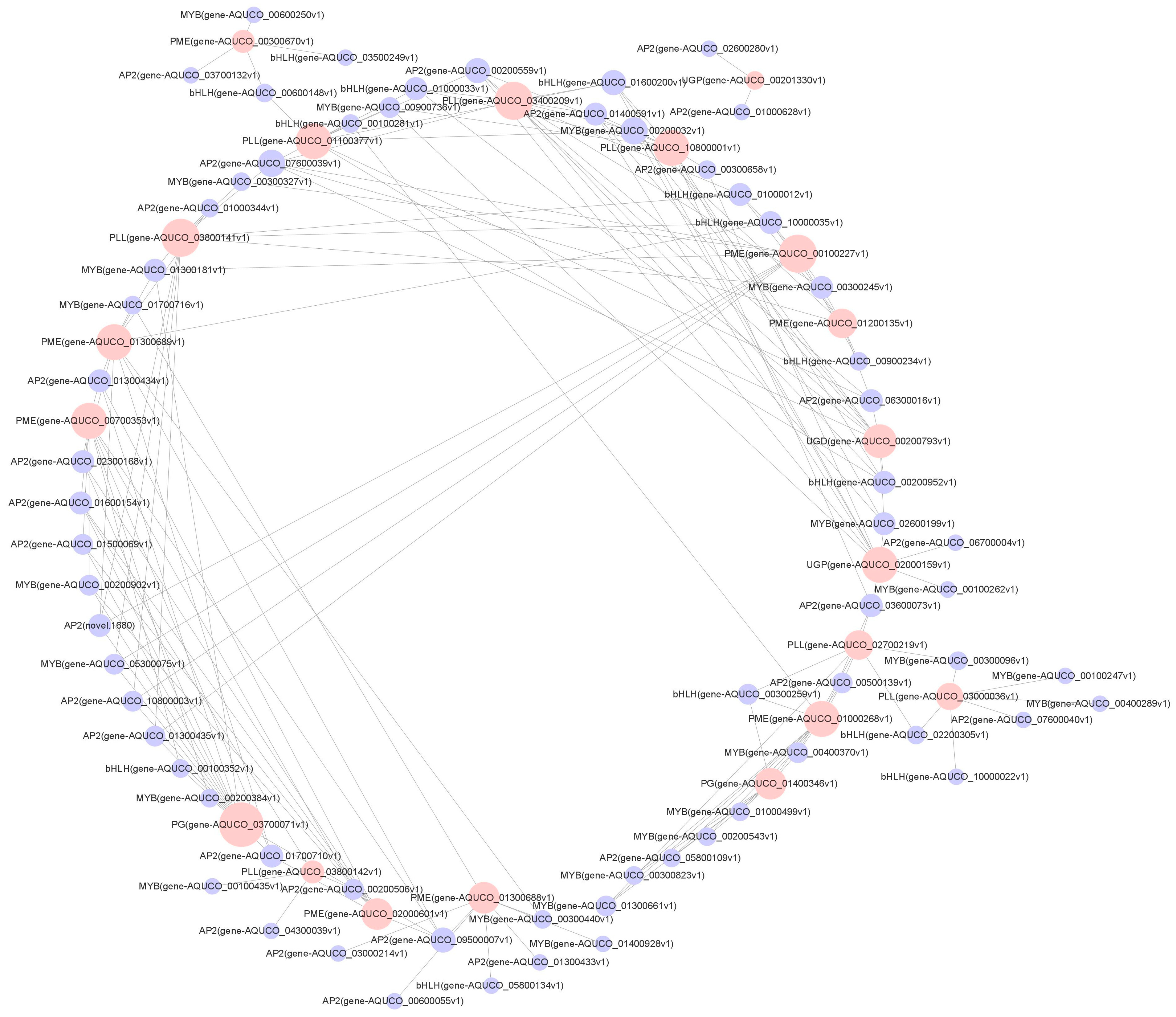
2.8. Network Analysis of Correlations between TFs and Functional Genes Related to the Cell Wall
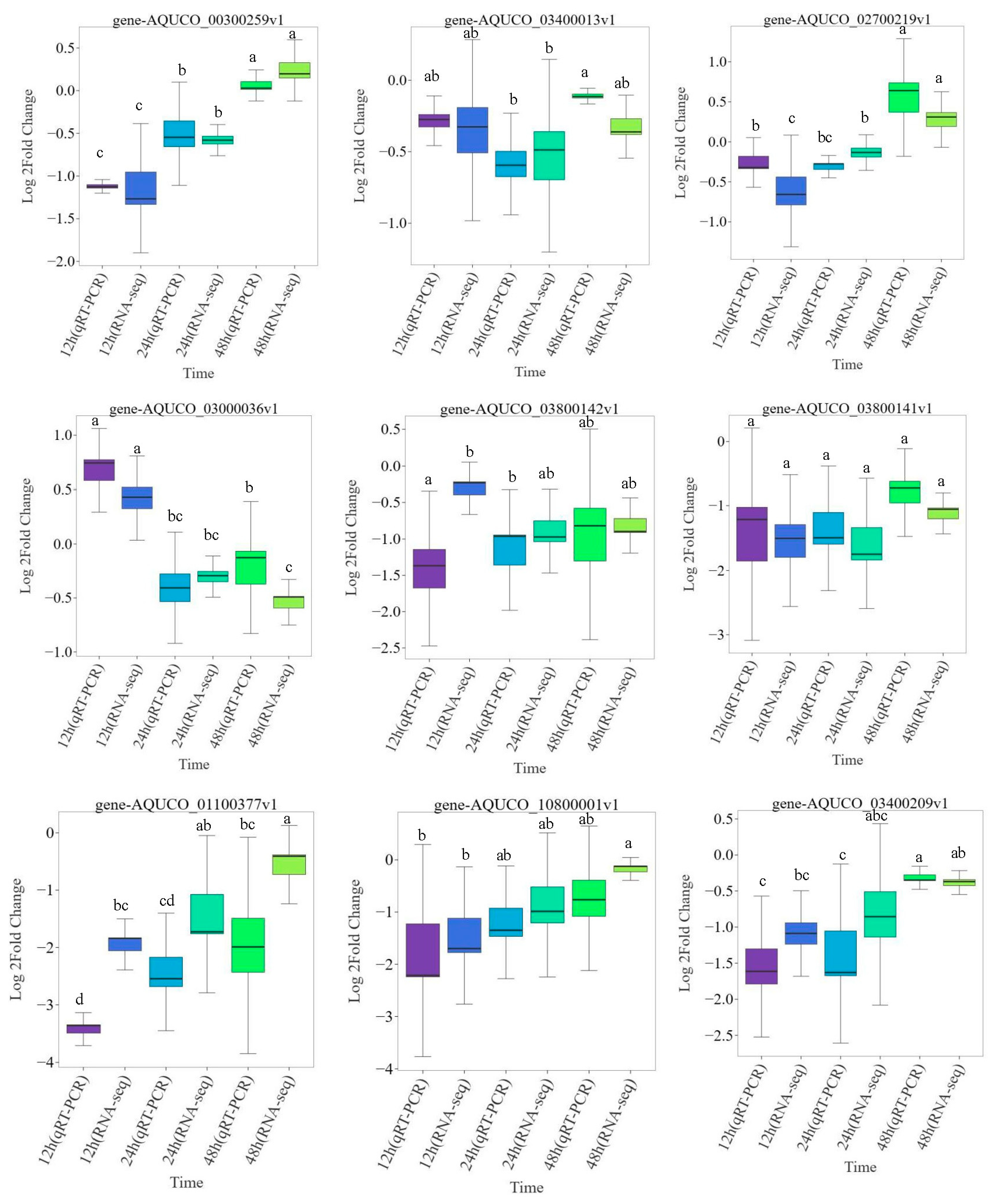
2.9. Validation of the RNA-Seq Data by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Treatment of Plant Material with Salt
4.2. Total RNA Extraction and cDNA Library Construction from the Roots of Aquilegia Vulgaris
4.3. Analysis of Transcriptome Data
4.4. Validation of Gene qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, M.; Liang, X. A HAK family Na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants 2019, 5, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Pang, J.Y. Salt-responsive transcriptome analysis of canola roots reveals candidate genes involved in the key metabolic pathway in response to salt stress. Sci. Rep. 2022, 12, 1666–1679. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.F.; Yu, J.H. Ionomic, metabolomic and proteomic analyses reveal molecular mechanisms of root adaption to salt stress in Tibetan wild barley. Plant Physiol. Biochem. 2018, 123, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.T.; Feng, S.J. Salt stressinduced FERROCHELATASE1 improves resistance to salt stress by limiting sodium accumulation in Arabidopsis thaliana. Sci. Rep. 2017, 7, 14737. [Google Scholar] [CrossRef]
- Zhang, X.X.; Liu, P. Comparative transcriptome analyses of maize seedling root responses to salt stress. PeerJ 2021, 9, 10765. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zhao, Y.J. Transcriptomic profiling of pomegranate provides insights into salt tolerance. Agron. J. 2019, 10, 44. [Google Scholar] [CrossRef]
- Sharmin, A.R.; Bhuiyan, R. RNA-Seq based transcriptomic analysis revealed genes associated with seed-flooding tolerance in wild soybean (Glycine soja Sieb. & Zucc.). Environ. Exp. Bot. 2020, 171, 103906. [Google Scholar] [CrossRef]
- Mo, S.Q.; Biao, A. Spatio transcriptome uncover novel insight into the Lycium ruthenicum seedling tolerant to salt stress. Ind. Crops Prod. 2022, 177, 114502. [Google Scholar] [CrossRef]
- Zhang, K.; Cui, H.T. Comparative time-course transcriptome analysis in contrasting Carex rigescens genotypes in response to high environmental salinity. Ecotoxicol. Environ. Saf. 2020, 194, 110435. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, K. Time-course transcriptomics analysis reveals key responses of populus to salt stress. Ind. Crops Prod. 2023, 194, 116278. [Google Scholar] [CrossRef]
- Dadshani, S.; Sharma, R.C. Multi-dimensional evaluation of response to salt stress in wheat. PLoS ONE 2019, 14, e0222659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Zhu, J.H. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 2, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018, 217, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Houston, K.; Tucker, M.R. The plant cell wall: A complex and dynamic structure as revealed by the responses of genes under stress conditions. Front. Plant Sci. 2016, 7, 984. [Google Scholar] [CrossRef]
- Lauri, V.; Julia, S. Cell wall integrity maintenance during plant development and interaction with the environment. Nat. Plants 2019, 5, 924–932. [Google Scholar] [CrossRef]
- Thorsten, H. The plant cell wall integrity maintenance mechanism—Concepts for organization and mode of action. Plant Cell Physiol. 2023, 56, 215–223. [Google Scholar] [CrossRef]
- Ding, A.M.; Tang, X.F. ERF4 and MYB52 transcription factors play antagonistic roles in regulating homogalacturonan de-methylesterification in Arabidopsis seed coat mucilage. Plant Cell 2020, 33, 381–403. [Google Scholar] [CrossRef]
- Li, J.B.; Sun, L.P. A stress-associated protein, PtSAP13, from Populus trichocarpa provides tolerance to salt stress. Int. J. Mol. Sci. 2019, 20, 5782. [Google Scholar] [CrossRef]
- Dai, W.S.; Wang, M. The transcription factor FcWRKY40 of Fortunella crassifolia functions positively in salt tolerance through modulation of ion homeostasis and proline biosynthesis by directly regulating SOS2 and P5CS1 homologs. New Phytol. 2018, 219, 972–989. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, R.L. The R2R3-MYB transcription factor AtMYB49 modulates salt tolerance in Arabidopsis by modulating the cuticle formation and antioxidant defence. Plant Cell Environ. 2020, 43, 1925–1943. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Vishal, B. Expression of AoNHX1 increases salt tolerance of rice and Arabidopsis, and bHLH transcription factors regulate AtNHX1 and AtNHX6 in Arabidopsis. Plant Cell Rep. 2019, 38, 1299–1315. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Luo, T.L. Identification of wheat DREB genes and functional characterizatio of TaDREB3 in response to abiotic stresses. Gene 2020, 740, e144514. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Meng, Y. Physio-biochemical responses of three Aquilegia species seedlings to salt stress. Agronomy 2022, 12, 2841. [Google Scholar] [CrossRef]
- Chen, L.F.; Meng, Y. Starch and sucrose metabolism and plant hormone signaling pathways play crucial roles in Aquilegia salt stress adaption. Int. J. Mol. Sci. 2023, 24, 3948. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Geng, F.D. Evidence for two types of Aquilegia ecalcarata and its implications for adaptation to new environments. Plant Divers. 2022, 44, 153–162. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.Y. Comparative chloroplast genomes and phylogenetic analysis of Aquilegia. Appl. Plant Sci. 2021, 9, e11412. [Google Scholar] [CrossRef]
- Edwards, M.B.; Choi, G. Genetic architecture of floral traits in bee- and hummingbird-pollinated sister species of Aquilegia (columbine). Evolution 2021, 75, 2197–2216. [Google Scholar] [CrossRef]
- Li, H.F.; Duijts, K. Effective root responses to salinity stress include maintained cell expansion and carbon allocation. New Phytol. 2023, 238, 1942–1956. [Google Scholar] [CrossRef]
- Deng, C.H.; Zhang, Z.B. Salt-responsive transcriptome analysis of triticale reveals candidate genes involved in the key metabolic pathway in response to salt stress. Sci. Rep. 2020, 10, e20669. [Google Scholar] [CrossRef]
- Cai, Z.C.; Wang, C.C. Comparative transcriptome analysis reveals variations of bioactive constituents in Lonicera japonica flowers under salt stress. Plant Physiol. Biochem. 2022, 173, 87–96. [Google Scholar] [CrossRef]
- Sun, S.Y.; Wang, Y.P. Transcriptome responses to salt stress in roots and leaves of Lilium pumilum. Sci. Hortic. 2023, 309, e111622. [Google Scholar] [CrossRef]
- Guerra, D.; Crosatti, C. Post-transcriptional and post-translational regulations of drought and heat response in plants: A spider’s web of mechanisms. Front. Plant Sci. 2015, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, H.L. Transcriptome analysis of two contrasting rice cultivars during alkaline stress. Sci. Rep. 2018, 8, 9586. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.L.; Dan, L. Transcriptome analysis of sugar beet (Beta vulgaris L.) in response to alkaline stress. Plant Mol. Biol. 2020, 102, 645–657. [Google Scholar] [CrossRef]
- Zhao, S.S.; Zhang, Q.K. Regulation of plant responses to salt stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.Y. Molecular insights into the complex mechanics of plant epidermal cell walls. Science 2021, 372, 706–711. [Google Scholar] [CrossRef]
- Endler, A.; Kesten, C. A mechanism for sustained cellulose synthesis during salt stress. Cell 2015, 162, 1353–1364. [Google Scholar] [CrossRef]
- Monniaux, M.; Hay, A. Cells, walls, and endless forms. Curr. Opin. Plant Biol. 2016, 34, 114–121. [Google Scholar] [CrossRef]
- Dinneny, J.R. Analysis of the salt-stress response at cell-type resolution. Plant Cell Environ. 2010, 33, 543–551. [Google Scholar] [CrossRef]
- Dai, Y.; Adnan, T.M. Physiological and transcriptomic response of soybean seedling roots to variable nitrate levels. Agron. J. 2021, 113, 3639–3652. [Google Scholar] [CrossRef]
- Lei, C.; Fan, S. Itraq-based proteomic analysis reveals potential regulation networks of iba-induced adventitious root formation in apple. Int. J. Mol. Sci. 2018, 19, 667. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Ye, C.T. Root hair single cell type specific profiles of gene expression and alternative polyadenylation under cadmium stress. Front. Plant Sci. 2019, 10, 589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, X.L. Identifying key regulatory genes of maize root growth and development by RNA sequencing. Genomics 2020, 112, 5157–5169. [Google Scholar] [CrossRef]
- Atmodjo, A.M.; Hao, Z.Y. Evolving views of pectin biosynthesis. Annu. Rev. Plant Biol. 2013, 64, 747–779. [Google Scholar] [CrossRef]
- Yan, J.W.; He, H. Pectin methylesterase31 positively regulates salt stress tolerance in Arabidopsis. Biochem. Biophys. Res. Commun. 2018, 496, 497–501. [Google Scholar] [CrossRef]
- Renault, H.; El-Amrani, A. γ-Aminobutyric acid transaminase deficiency impairs central carbon metabolism and leads to cell wall defects during salt stress in Arabidopsis roots. Plant Cell Environ. 2013, 36, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.L.; Liu, H.J. Molecular evolution and expression divergence of the Populus polygalacturonase supergene family shed light on the evolution of increasingly complex organs in plants. New Phytol. 2013, 197, 1353–1365. [Google Scholar] [CrossRef]
- Yan, S.P.; Tang, Z.C. Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 2005, 5, 235–244. [Google Scholar] [CrossRef]
- Mostek, A.; Börner, A. Alterations in root proteome of salt-sensitive and tolerant barley lines under salt stress conditions. Plant Physiol. 2015, 17, 166–176. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, L. Membrane lipid remodeling in response to salinity. Int. J. Mol. Sci. 2019, 20, 4264. [Google Scholar] [CrossRef]
- Zhang, B.C.; Gao, Y.H. The plant cell wall: Biosynthesis, construction, and functions. Integr. Plant Biol. 2021, 63, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.P.; Tian, F. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Yang, W.J. TaMYB86B encodes a R2R3-type MYB transcription factor and enhances salt tolerance in wheat. Plant Sci. 2020, 300, 110624. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Liu, B. LpNAC6 reversely regulates the alkali tolerance and drought tolerance of Lilium pumilum. Plant Physiol. 2022, 270, 153635. [Google Scholar] [CrossRef]
- Xie, R.J.; Pan, X.T. Effect of salt-stress on gene expression in citrus roots revealed by RNA-seq. Funct. Integr. Genom. 2018, 18, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.D.; Jiang, Y.L. Expression of the maize MYB transcription factor ZmMYB3R enhances drought and salt stress tolerance in transgenic plants. Plant Physiol. Biochem. 2019, 137, 179–188. [Google Scholar] [CrossRef]
- Sun, H.R.; Hao, P.B. Pectate lyase-like Gene GhPEL76 regulates organ elongation in Arabidopsis and fiber elongation in cotton. Plant Sci. 2020, 293, 110395. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Garber, M.; Grabherr, M.G. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, W.W. Transcriptome analysis reveals different response of resistant and susceptible rice varieties to rice stripe virus infection. J. Integr. Agric. 2023, 22, 1750. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G. clusterProfiler: An R package for comparing biological themes among gene clusters. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K. Feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Li, X.; Gao, J. Multi-omics analyses of 398 foxtail millet accessions reveal genomic regions associated with domestication, metabolite traits, and anti-inflammatory effects. Mol. Plant 2022, 15, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Kramer, E. Sub- and neo-functionalization of APETALA3 paralogs have contributed to the evolution of novel floral organ identity in Aquilegia (columbine, Ranunculaceae). New Phytol. 2013, 197, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Yu, H.; Chen, L.; Meng, Y.; Ma, Y.; Wang, D.; Qian, Y.; Zhang, D.; Feng, X.; Zhou, Y. Time-Course Transcriptome Analysis of Aquilegia vulgaris Root Reveals the Cell Wall’s Roles in Salinity Tolerance. Int. J. Mol. Sci. 2023, 24, 16450. https://doi.org/10.3390/ijms242216450
Bai Y, Yu H, Chen L, Meng Y, Ma Y, Wang D, Qian Y, Zhang D, Feng X, Zhou Y. Time-Course Transcriptome Analysis of Aquilegia vulgaris Root Reveals the Cell Wall’s Roles in Salinity Tolerance. International Journal of Molecular Sciences. 2023; 24(22):16450. https://doi.org/10.3390/ijms242216450
Chicago/Turabian StyleBai, Yun, Haihang Yu, Lifei Chen, Yuan Meng, Yanmei Ma, Di Wang, Ying Qian, Dongyang Zhang, Xiaoyu Feng, and Yunwei Zhou. 2023. "Time-Course Transcriptome Analysis of Aquilegia vulgaris Root Reveals the Cell Wall’s Roles in Salinity Tolerance" International Journal of Molecular Sciences 24, no. 22: 16450. https://doi.org/10.3390/ijms242216450