
An Unsupervised Classifier for Whole-Genome Phylogenies, the Maxwell© Tool

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biological Context
2.1. Gene Translation for Building Proteins
2.2. Combinatorial Properties of AL
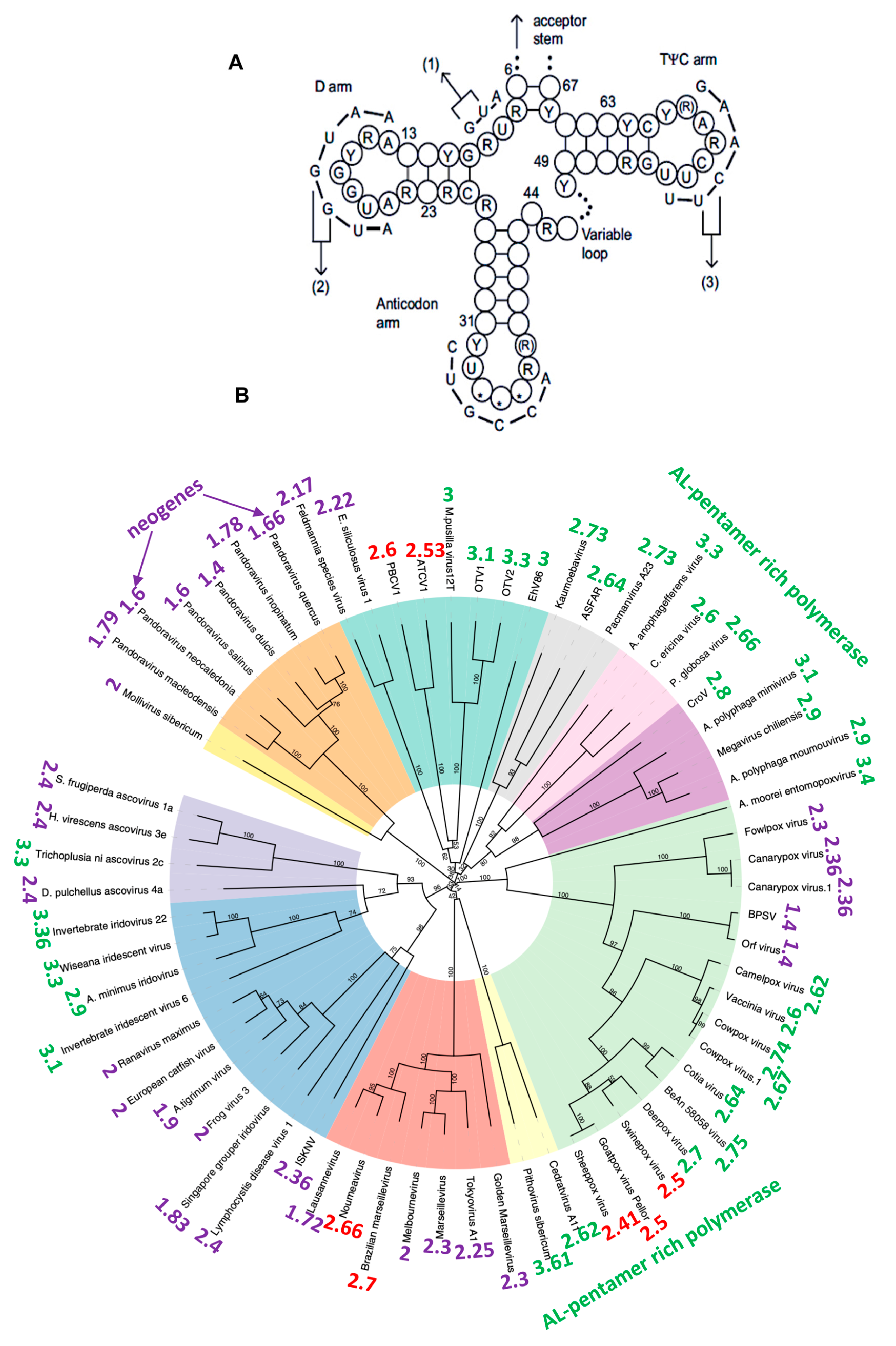
2.3. Traces of the Archetypal Loop AL in the Present Genomes
2.4. Ribosomal Proteins and rRNA Components of the Current Ribosomes
3. Results
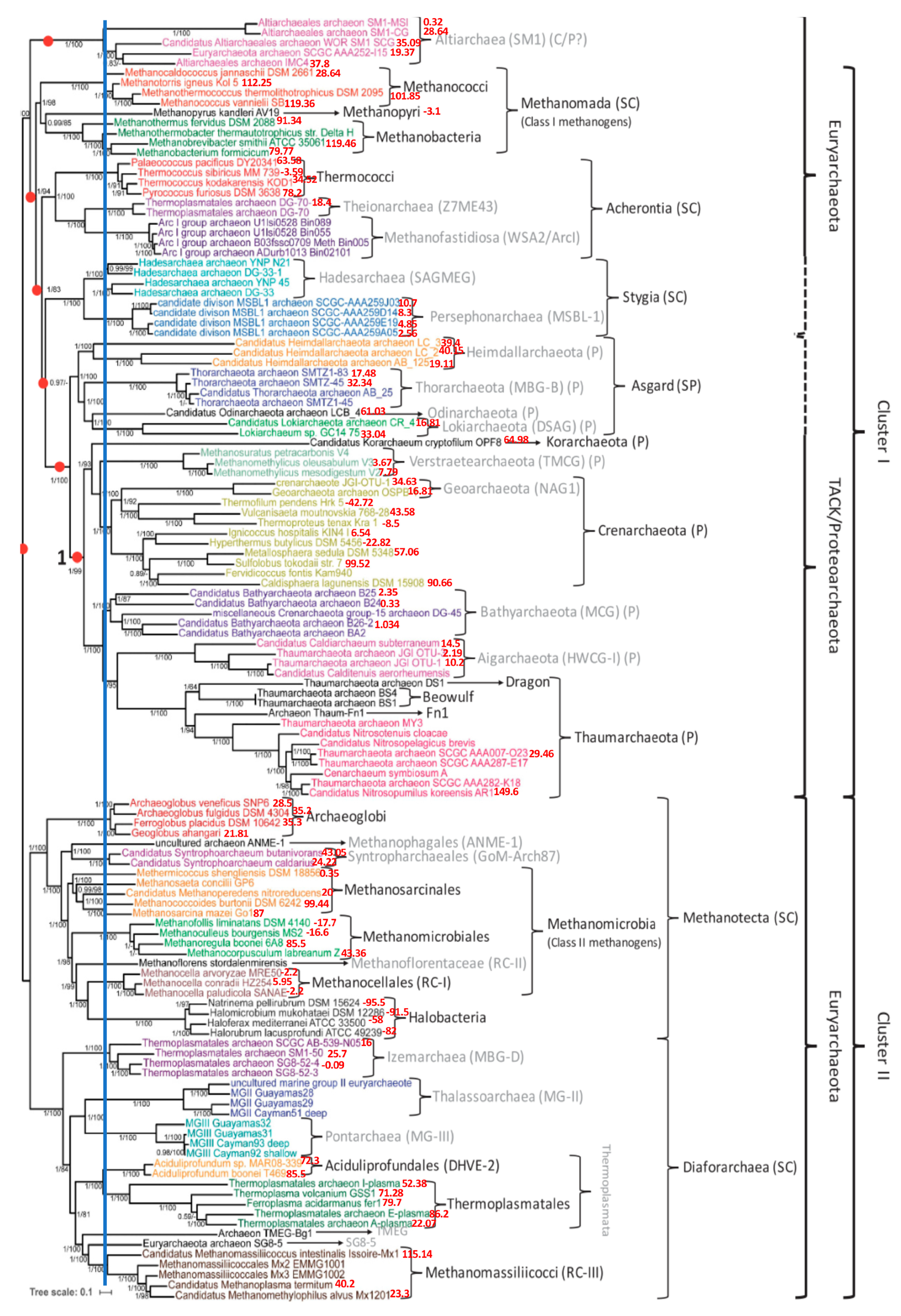
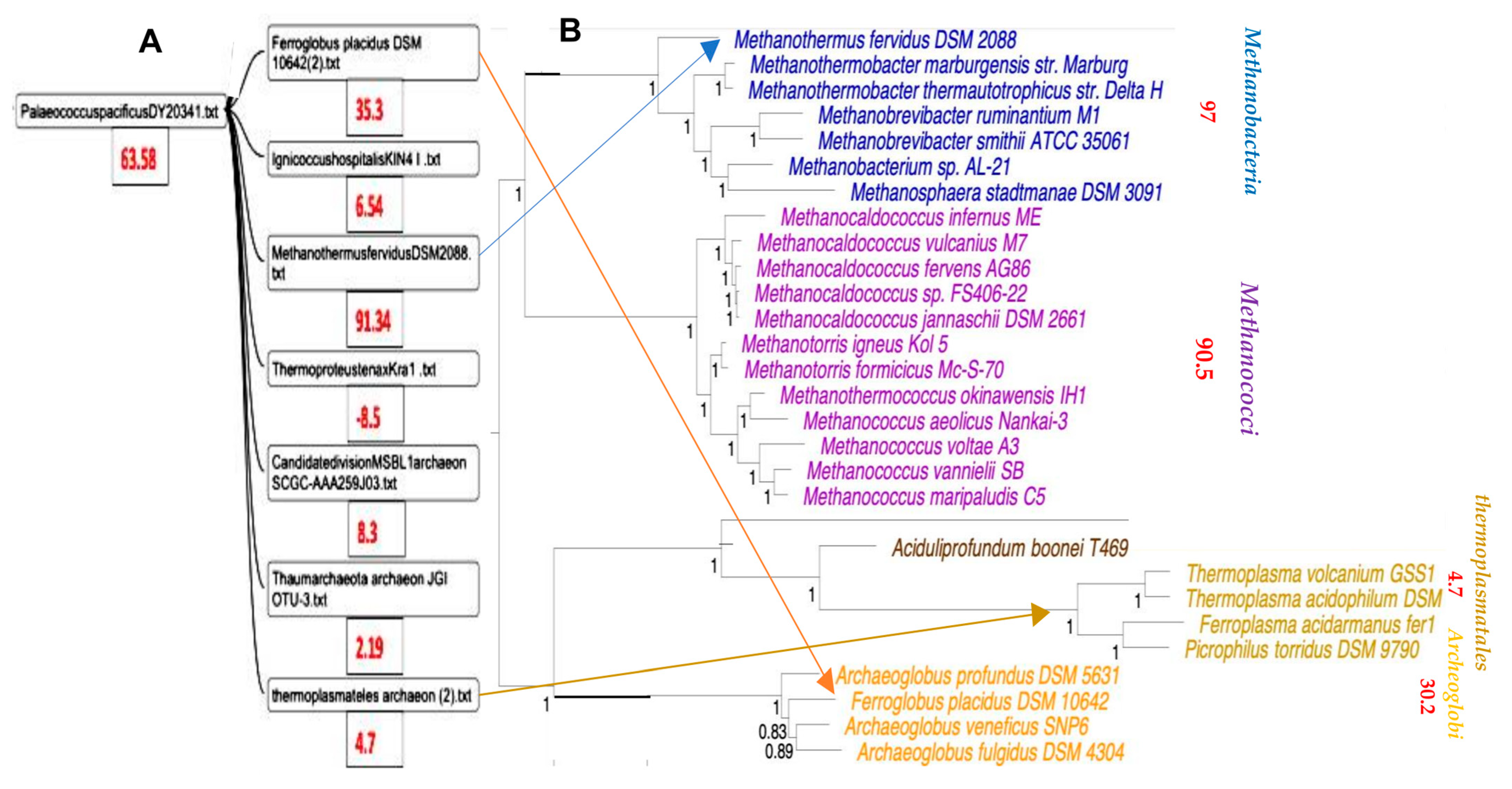
3.1. Phylogenetic Tree of Archaea
3.2. Classification of Archaea by Maxwell
4. Discussion
5. Methodology
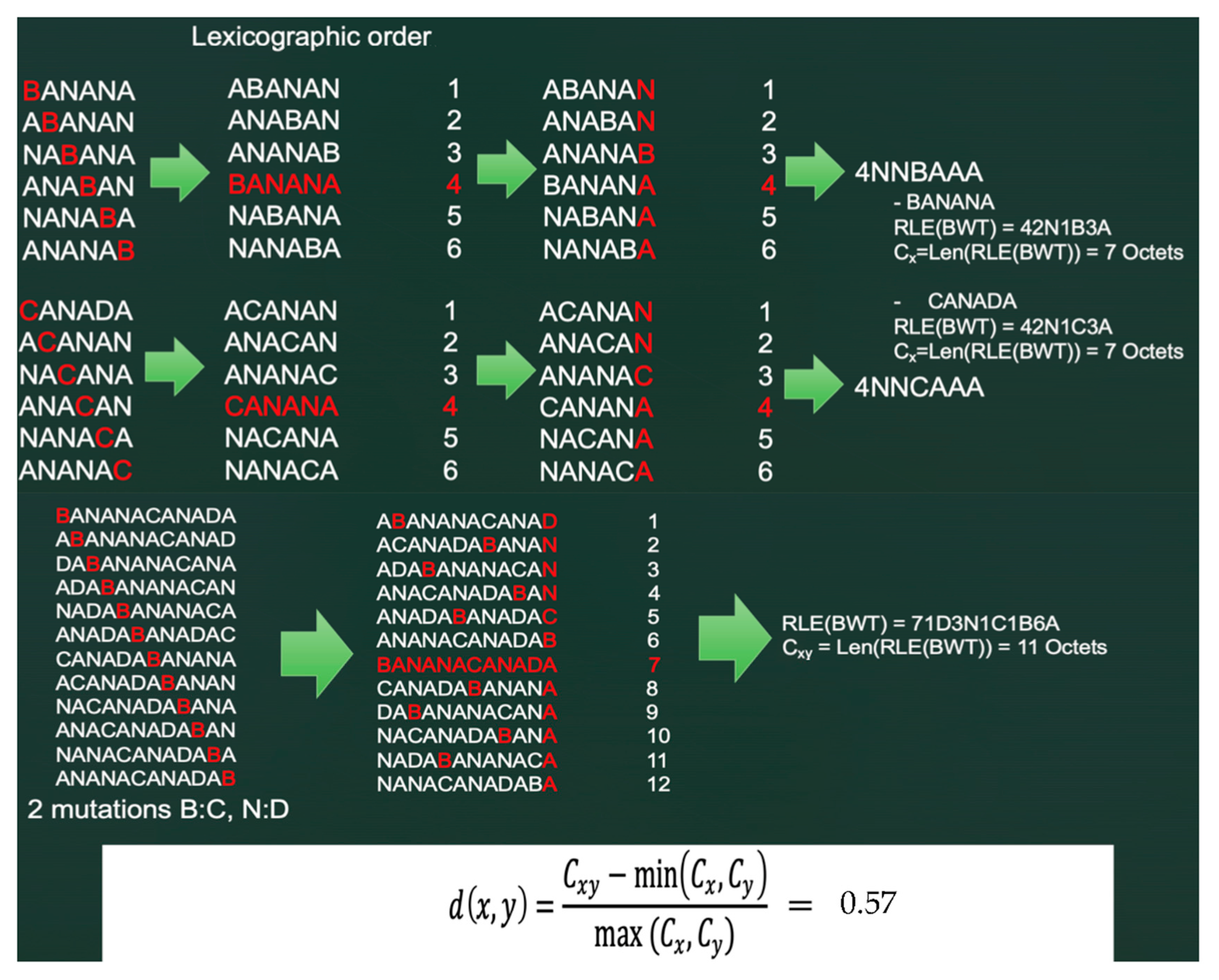
5.1. The Burrows–Wheeler Transform
5.2. Principles of the Burrows–Wheeler Transform (BWT)
5.3. “Normalized Compression Distance” (NCD) or Vitányi Distance
5.4. Steps of the Maxwell© Algorithm of Clustering (MAC)
- (1)
- One-shot learning. This procedure is static. It includes a unique forward pass whose role is to calculate the similarity clusters.
- (2)
- Active learning. This procedure is dynamic. It includes one or several cycles of forward–backward passes. A backward pass consists of adjusting the validity of the clusters calculated previously in the forward pass based on a feedback loop. Feedback can be carried out either through automated decision, based on a validity calculation, or through the intervention of a human expert decision. At the end of a forward–backward cycle, invalidated data and/or clusters are rejected as singleton to possibly be then incorporated into a new forward–backward learning cycle, which can be summarized as follows:
- (1)
- Forward pass
- -
- The forward pass is the core of the original algorithm of Maxwell©. It takes as input the distance matrix Dij and the triangulation standard deviation parameter σ to then execute the following steps:
- -
- Triangulation property calculation: calculation of mean and standard deviation on histograms of triangle areas for filtering “large and deformed triangles” considered as outliers of the empirical distribution, according to the observed number of standard deviations σ from the area mean value retained;
- -
- First-of-first (1-1) neighborhood pruning: examining sub-graphs whose “useless” (respectively, “best”) representative edges are identified as attached to the least (respectively, the most) connected nodes and remove (respectively, keep) them as central nodes;
- -
- Local minima detection: processing subgraphs with several local minima, i.e., nodes whose neighborhood does not contain another node that is closer to the subgraph than the node itself, by using Voronoï networking (as in [5]) for detecting internal boundaries. In practice, this step can be implemented using the Graphiz open API [39] by testing at the end for sub-graphs of which mean and standard deviation depend on local minima, until Graphviz no longer detects any internal boundary;
- -
- Singleton formation: storage of all the elements rejected by this statistical calculation in the form of “singleton clusters”;
- -
- Final recall: re-clustering the population of singletons to detect new clusters.
- (2)
- Backward pass
5.5. Toward Auto-Correction in Maxwell© with Multisets Used for Best Representative Nodes
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steinhaus, H. Sur la division des corps matériels en parties. Bull. Acad. Polon. Sci. 1957, 4, 801–804. [Google Scholar]
- MacQueen, J.B. Some Methods for classification and Analysis of Multivariate Observations. Proceedings of 5th Berkeley Symposium on Mathematical Statistics and Probability, Berkeley, CA, USA, 21 June–18 July 1965 and 27 December 1965–7 January 1966; University of California Press: Berkeley, CA, USA, 1967; Volume 1, pp. 281–297. [Google Scholar]
- Diday, E. Une nouvelle méthode en classification automatique et reconnaissance des formes la méthode des nuées dynamiques. Rev. Stat. Appl. 1971, 19, 19–33. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Gualtieri, J.A.; Cromp, R.F. Support vector machines for hyperspectral remote sensing classification. Proc. SPIE 1998, 3584, 221–232. [Google Scholar]
- Hopfield, J.J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef]
- Mattes, J.; Demongeot, J. Dynamic confinement, classification and imaging. In Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 1999; Volume 14, pp. 205–214. [Google Scholar]
- Demongeot, J.; Sené, S. The singular power of the environment on nonlinear Hopfield networks. In CMSB’11, ACM Proceedings; ACM: New York, NY, USA, 2011; pp. 55–64. [Google Scholar]
- Hinton, G. A Practical Guide to Training Restricted Boltzmann Machines. In Neural Networks: Tricks of the Trade; Lecture Notes in Computer Science Series; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7700, pp. 599–619. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Cox, A.J.; Bauer, M.J.; Jakobi, T.; Rosone, G. Large-scale compression of genomic sequence databases with the Burrows-Wheeler transform. Bioinformatics 2012, 28, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- tRNAviz. Available online: http://trna.ucsc.edu/tRNAviz/ (accessed on 23 May 2023).
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Demongeot, J.; Moreira, A. A circular RNA at the origin of life. J. Theor. Biol. 2007, 249, 314–324. [Google Scholar] [CrossRef]
- Rigden, D.J.; Fernández, X.M. The 2021 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 2021, 49, D1–D9. [Google Scholar] [CrossRef]
- Lee, B.D.; Neri, U.; Oh, C.J.; Simmonds, P.; Koonin, E.V. ViroidDB: A database of viroids and viroid-like circular RNAs. Nucleic Acids Res. 2022, 50, D432–D438. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Raoult, D. Stem-Loop RNA Hairpins in Giant Viruses: Invading rRNA-Like Repeats and a Template Free RNA. Front. Microbiol. 2018, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C. Prebiotic RNA Engineering in a Clay Matrix and the Origin of Life: Mechanistic and Molecular Modeling Rationale for Explaining the Helicity, Antiparallelism and Prebiotic Replication of Nucleic Acids. BME Horiz. 2023. to appear. [Google Scholar]
- Demongeot, J.; Seligmann, H. Spontaneous evolution of circular codes in theoretical minimal RNA rings. Gene 2019, 705, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Demongeot, J.; Gardes, J.; Maldivi, C.; Boisset, D.; Boufama, K.; Touzouti, I. Genomic phylogeny by Maxwell®, a new classifier based on Burrows-Wheeler transform. Computation 2023, 11, 158. [Google Scholar] [CrossRef]
- Demongeot, J.; Thellier, M. Primitive oligomeric RNAs at the origins of life on Earth. Int. J. Mol. Sci. 2023, 24, 2274. [Google Scholar] [CrossRef] [PubMed]
- Novozhilov, A.S.; Koonin, E.V. Exceptional error minimization in putative primordial genetic codes. Biol. Direct. 2009, 4, 44. [Google Scholar] [CrossRef]
- Trifonov, E.N. Consensus temporal order of amino acids and evolution of the triplet code. Gene 2000, 261, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Harish, A.; Caetano-Anollés, G. Ribosomal History Reveals Origins of Modern Protein Synthesis. PLoS ONE 2012, 7, e32776. [Google Scholar] [CrossRef]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 23 June 2023).
- Luk, A.W.; Williams, T.J.; Erdmann, S.; Papke, R.T.; Cavicchioli, R. Viruses of haloarchaea. Life 2014, 4, 681–715. [Google Scholar] [CrossRef]
- Ngo, V.Q.H.; Enault, F.; Midoux, C.; Mariadassou, M.; Chapleur, O.; Mazéas, L.; Loux, V.; Bouchez, T.; Krupovic, M.; Bize, A. Diversity of novel archaeal viruses infecting methanogens discovered through coupling of stable isotope probing and metagenomics. Env. Microbiol. 2022, 24, 4853–4868. [Google Scholar] [CrossRef] [PubMed]
- Matte-Tailliez, O.; Brochier, C.; Forterre, P.; Philippe, H. Archaeal phylogeny based on ribosomal proteins. Mol. Biol. Evol. 2002, 19, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, C.; Deschamps, P.; López-García, P.; Moreira, D.; Brochier-Armanet, C. Extending the conserved phylogenetic core of archaea disentangles the evolution of the third domain of life. Mol. Biol. Evol. 2015, 32, 1242–1254. [Google Scholar] [CrossRef]
- Tahon, G.; Geesink, P.; Ettema, T.J.G. Expanding Archaeal Diversity and Phylogeny: Past, Present, and Future. Annu. Rev. Microbiol. 2021, 75, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L. Directionality Theory and the Origin of Life. arXiv 2023, arXiv:2304.14873. [Google Scholar]
- Gardes, J.; Maldivi, C.; Boisset, D.; Aubourg, T.; Vuillerme, N.; Demongeot, J. Maxwell®: An unsupervised learning approach for 5P medicine. Stud. Health Technol. Inform. 2019, 264, 1464–1465. [Google Scholar]
- Burrows, M.; Wheeler, D.J. A block-sorting lossless data compression algorithm. Digit. SRC Res. Rep. 1994, 124, 1–24. [Google Scholar]
- Royer, L.; Reimann, M.; Andreopoulos, B.; Schroeder, M. Unraveling Protein Networks with Power Graph Analysis. PLoS Comput. Biol. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Agustsson, E.; Mentzer, F.; Tschannen, M.; Cavigelli, L.; Timofte, R.; Benini, L.; Gool, L.V. Soft-to-Hard Vector Quantization for End-to-End Learning Compressible Representations. Adv. Neural Inf. Process. Syst. 2017, 30, 1141–1151. [Google Scholar]
- Cilibrasi, R.; Vitányi, P.M.B. Clustering by compression. IEEE Trans. Inf. Theory 2005, 51, 1523–1545. [Google Scholar] [CrossRef]
- Cohen, A.R.; Vitányi, P.M.B. Normalized Compression Distance of Multisets with Applications. IEEE Trans. PAMI 2015, 37, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Graphviz. Available online: https://graphviz.org/ (accessed on 23 May 2023).
- Vardasbi, A.; Faili, H.; Asadpour, M. On the Reselection of Seed Nodes in Independent Cascade Based Influence Maximization. Int. J. Inf. Commun. Technol. Res. 2018, 10, 11–21. [Google Scholar]
- Pastor-Satorras, R.; Castellano, C.; Van Mieghem, P.; Vespignani, A. Epidemic processes in complex networks. Rev. Mod. Phys. 2015, 87, 925–979. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardes, J.; Maldivi, C.; Boisset, D.; Aubourg, T.; Demongeot, J. An Unsupervised Classifier for Whole-Genome Phylogenies, the Maxwell© Tool. Int. J. Mol. Sci. 2023, 24, 16278. https://doi.org/10.3390/ijms242216278
Gardes J, Maldivi C, Boisset D, Aubourg T, Demongeot J. An Unsupervised Classifier for Whole-Genome Phylogenies, the Maxwell© Tool. International Journal of Molecular Sciences. 2023; 24(22):16278. https://doi.org/10.3390/ijms242216278
Chicago/Turabian StyleGardes, Joël, Christophe Maldivi, Denis Boisset, Timothée Aubourg, and Jacques Demongeot. 2023. "An Unsupervised Classifier for Whole-Genome Phylogenies, the Maxwell© Tool" International Journal of Molecular Sciences 24, no. 22: 16278. https://doi.org/10.3390/ijms242216278