
RNF138 Downregulates Antiviral Innate Immunity by Inhibiting IRF3 Activation
 and
and 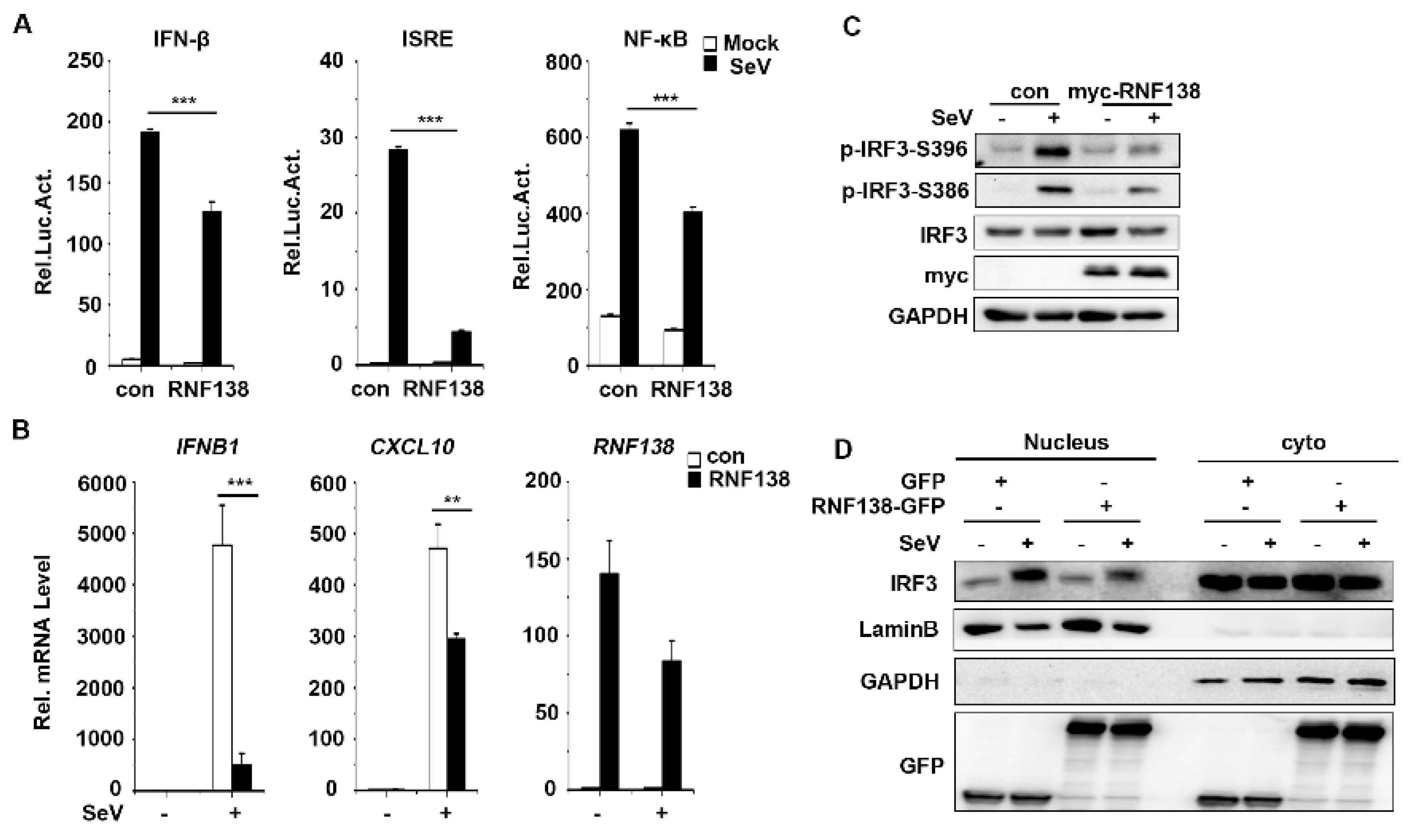{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RNF138 Negatively Regulates Viral RNA-Triggered Signaling
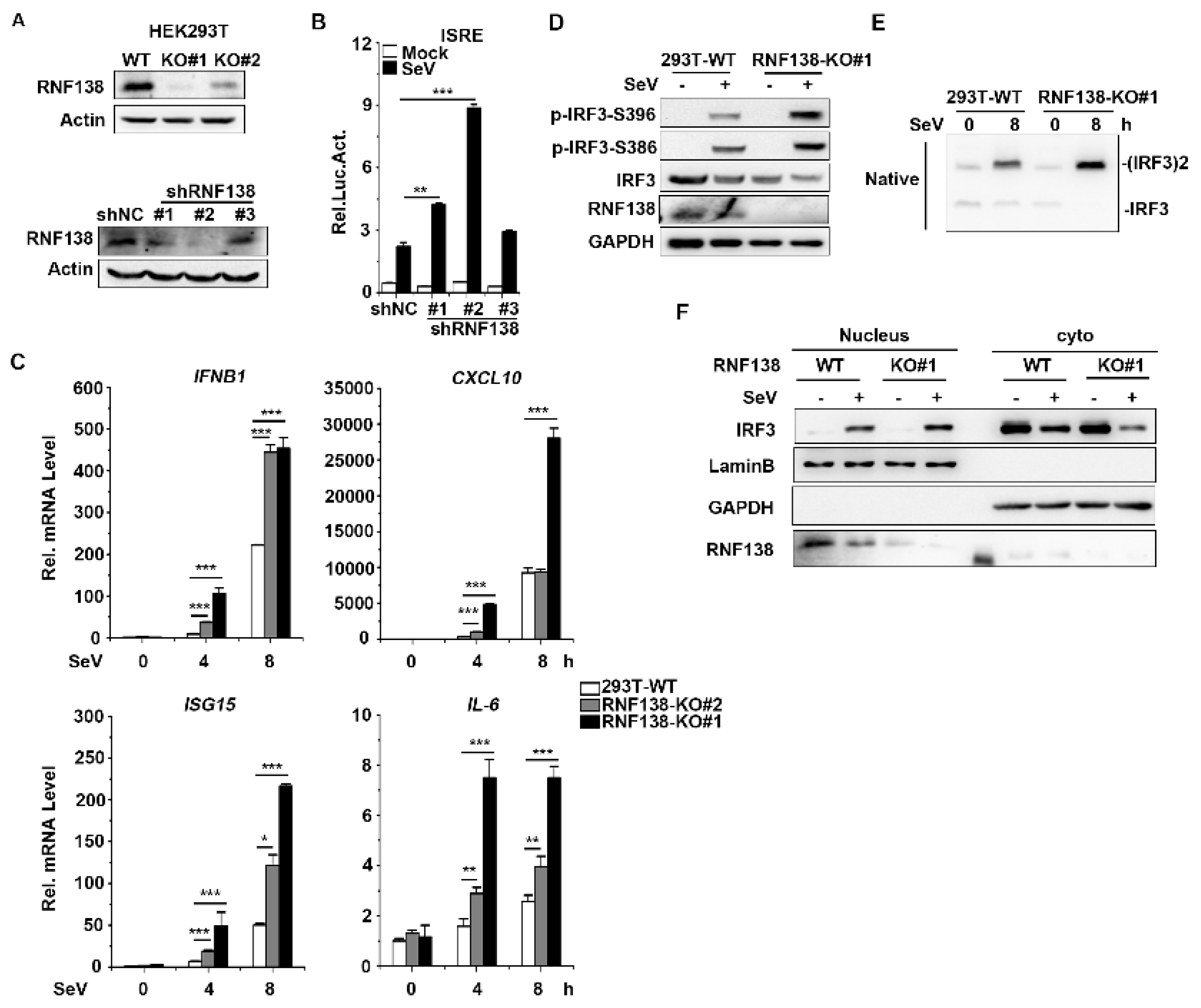
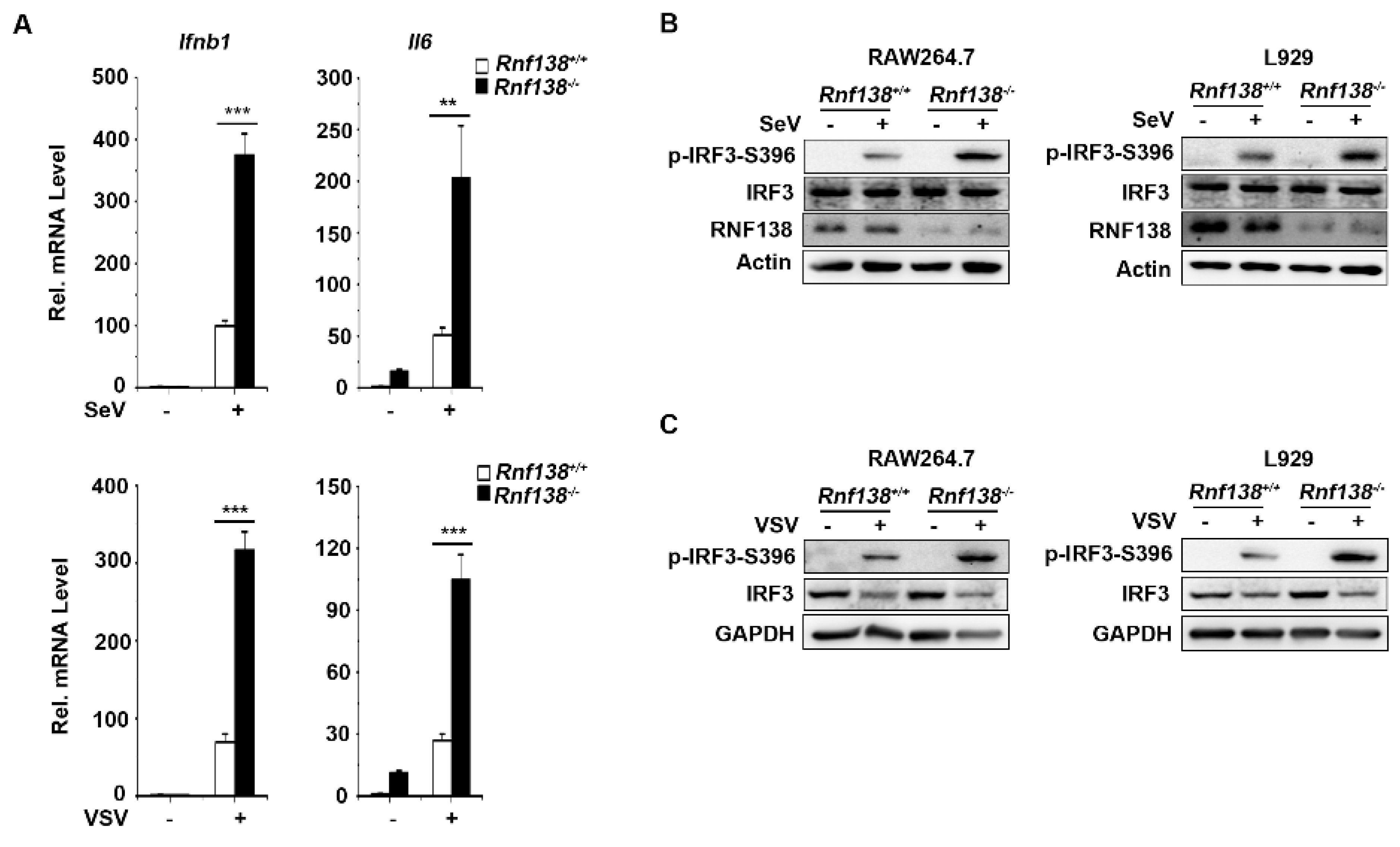
2.2. Knockout of RNF138 Potentiates Virus-Induced IRF3 Activation and IFNB1 Transcription
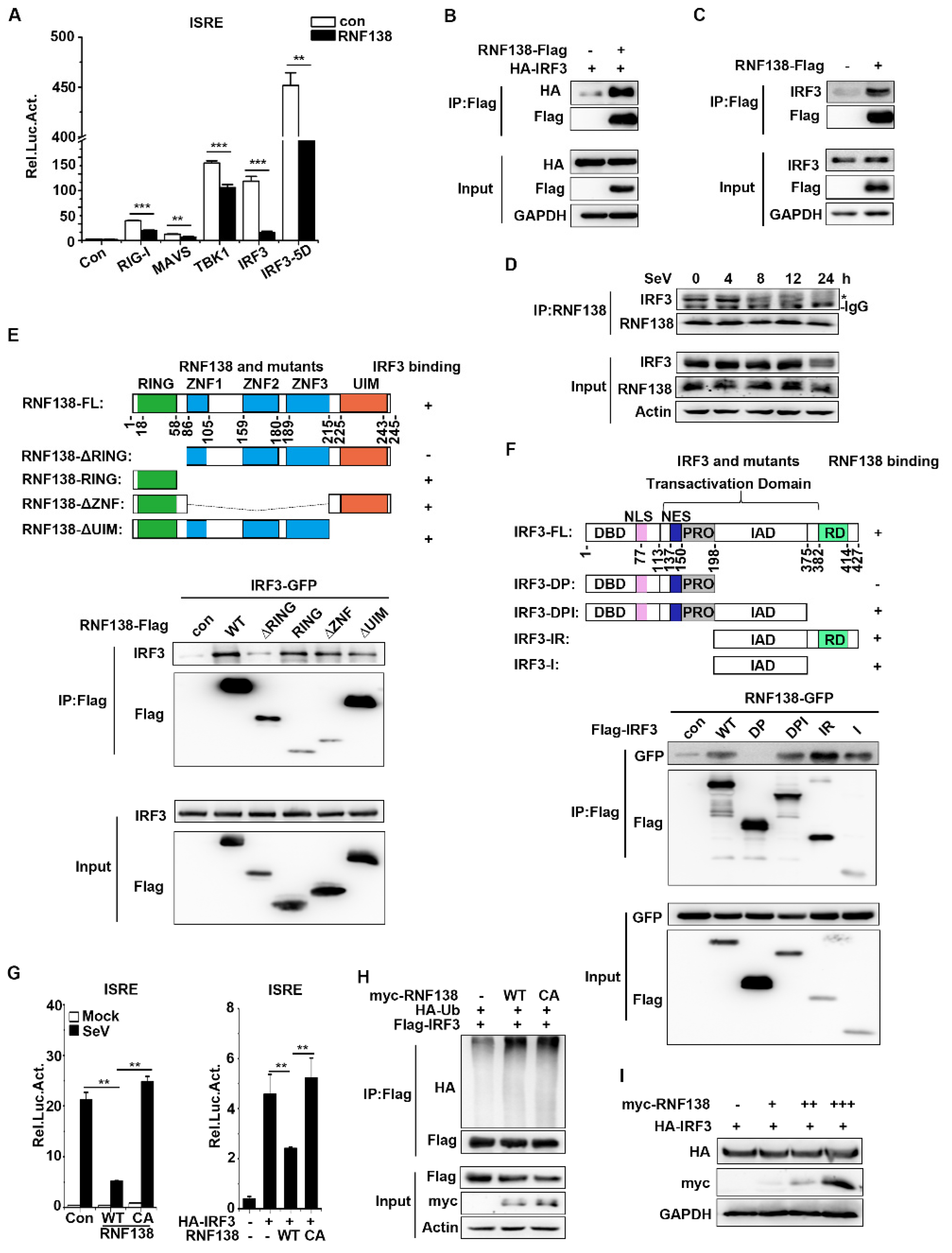
2.3. RNF138 Negatively Regulates Virus-Triggered Signaling by Targeting IRF3
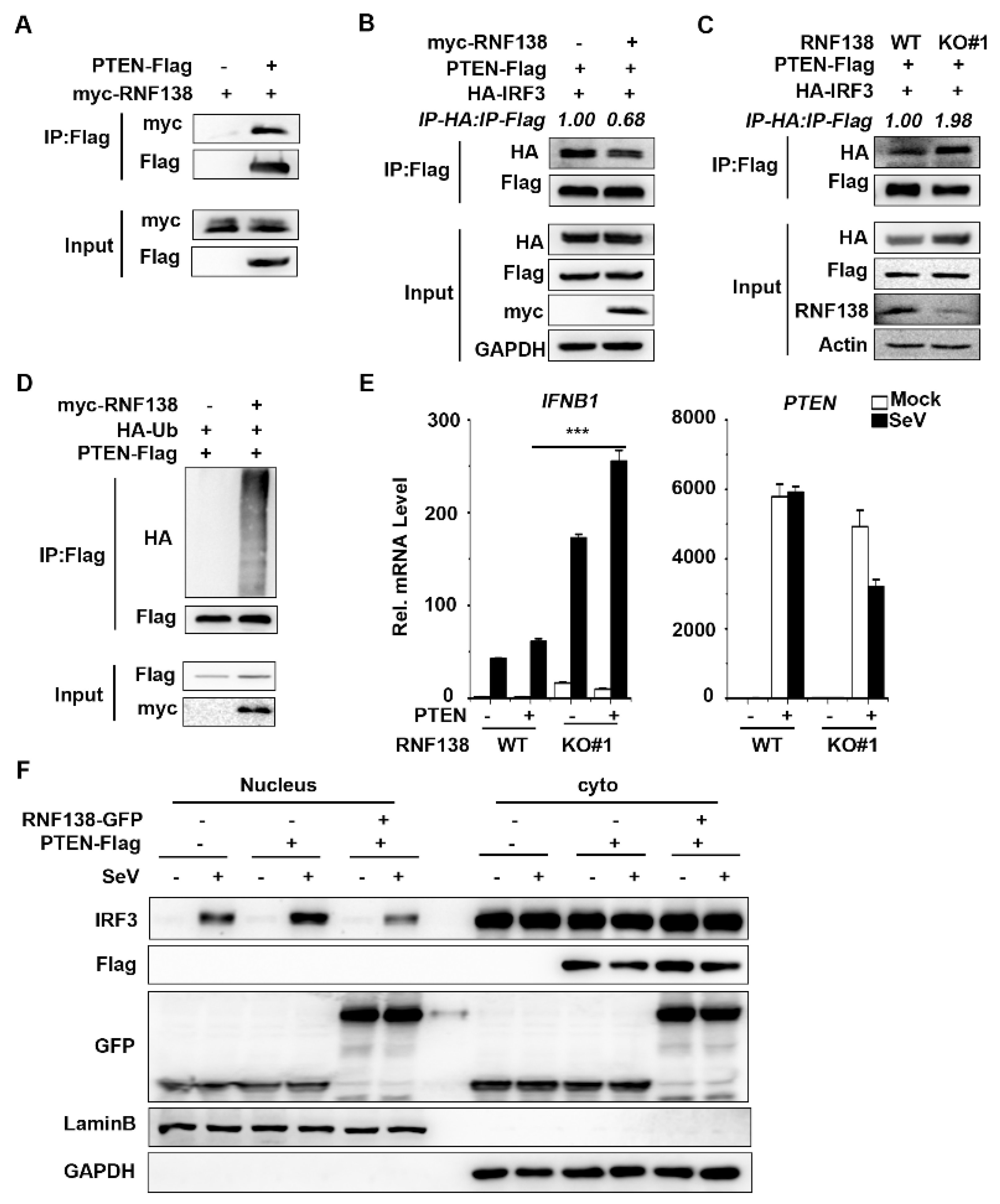
2.4. RNF138 Inhibits IRF3 Activation by Ubiquitinating PTEN
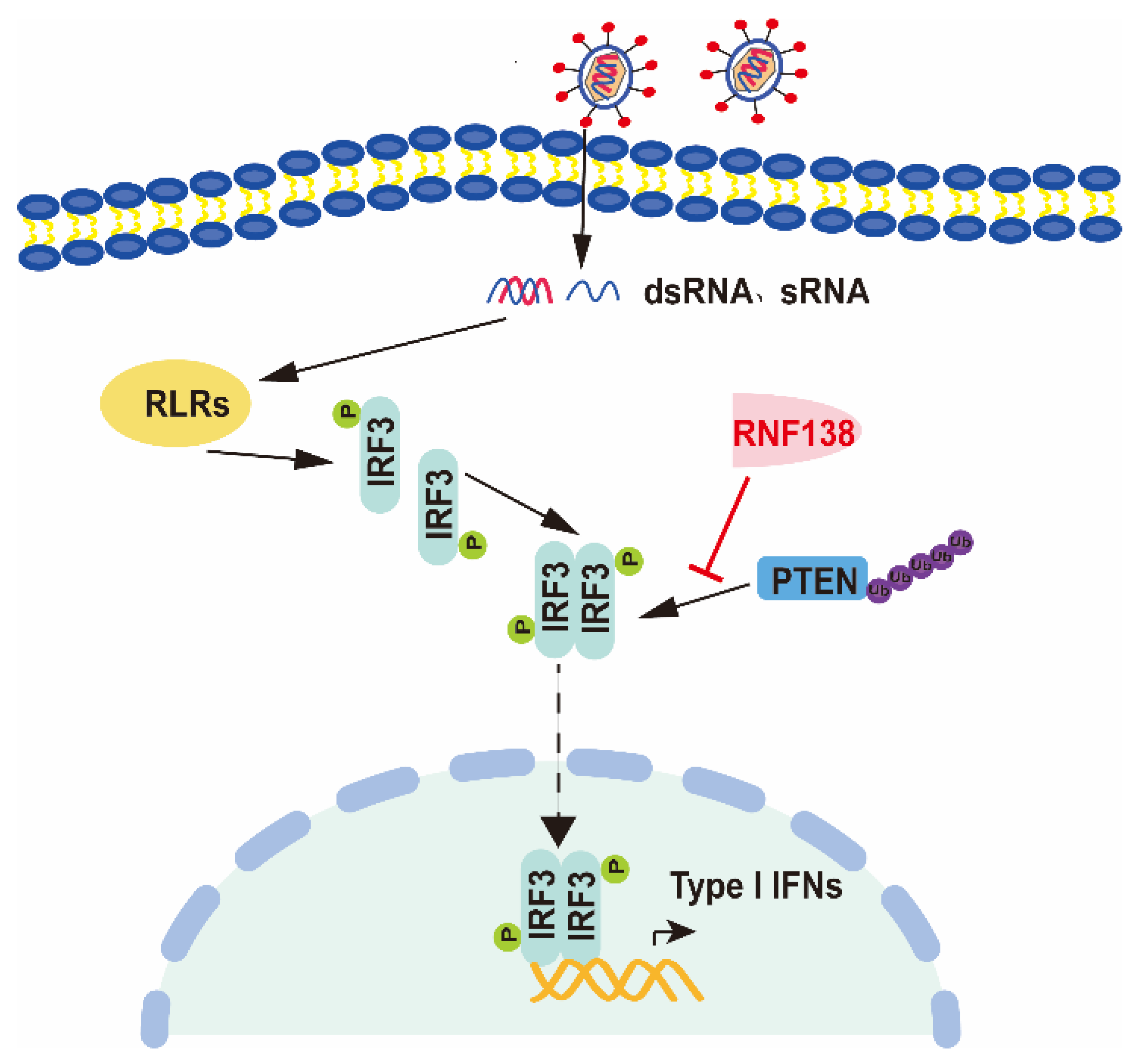
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Constructs
4.3. Transfection and Reporter Gene Assays
4.4. PCR
4.5. shRNA
4.6. Generation of Knockout Cells by CRISPR/Cas9 Technology
4.7. Co-Immunoprecipitation and Immunoblotting Analyses
4.8. Ubiquitination Assays
4.9. Native PAGE
4.10. Subcellular Fractionation
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Hu, M.M.; Shu, H.B. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annu. Rev. Cell Dev. Biol. 2018, 34, 357–379. [Google Scholar] [CrossRef]
- Zou, J.; Kawai, T.; Tsuchida, T.; Kozaki, T.; Tanaka, H.; Shin, K.S.; Kumar, H.; Akira, S. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity 2013, 38, 717–728. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Sousa, C.R.E. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Chuenchor, W.; Jin, T.; Ravilious, G.; Xiao, T.S. Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr. Opin. Immunol. 2014, 26, 14–20. [Google Scholar] [CrossRef]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 2008, 60, 805–812. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Yip, T.F.; Yan, S.; Jin, D.Y.; Wei, H.L.; Guo, R.T.; Peiris, J.S.M. Recognition of Double-Stranded RNA and Regulation of Interferon Pathway by Toll-Like Receptor 10. Front. Immunol. 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998, 425, 112–116. [Google Scholar] [CrossRef]
- Qin, B.Y.; Liu, C.; Lam, S.S.; Srinath, H.; Delston, R.; Correia, J.J.; Derynck, R.; Lin, K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 2003, 10, 913–921. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Yoneyama, M.; Ito, T.; Takahashi, K.; Inagaki, F.; Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 2004, 279, 9698–9702. [Google Scholar] [CrossRef] [PubMed]
- Servant, M.J.; Grandvaux, N.; tenOever, B.R.; Duguay, D.; Lin, R.; Hiscott, J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 2003, 278, 9441–9447. [Google Scholar] [CrossRef]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, M.X.; Tang, Z.; Zhang, Q.; Ye, J.; Xiong, T.C.; Zhang, Z.D.; Zhong, B. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J. Exp. Med. 2020, 217, e20191174. [Google Scholar] [CrossRef]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.Y.; Chen, S.; Zhao, X.; Lei, C.Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef]
- Ismail, I.H.; Gagne, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat. Cell Biol. 2015, 17, 1458–1470. [Google Scholar] [CrossRef]
- Xu, L.; Lu, Y.; Han, D.; Yao, R.; Wang, H.; Zhong, S.; Luo, Y.; Han, R.; Li, K.; Fu, J.; et al. Rnf138 deficiency promotes apoptosis of spermatogonia in juvenile male mice. Cell Death Dis. 2017, 8, e2795. [Google Scholar] [CrossRef]
- Yu, X.; Li, W.; Deng, Q.; Liu, H.; Wang, X.; Hu, H.; Cao, Y.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-κB activation and lymphomagenesis. Blood 2021, 137, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Byun, K.; Hong, W.; Chuang, H.Y.; Pack, C.G.; Bayarsaikhan, E.; Paek, S.H.; Kim, H.; Shin, H.Y.; Ideker, T.; et al. Proteome-wide discovery of mislocated proteins in cancer. Genome Res. 2013, 23, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Long, P.; Samnakay, P.; Jenner, P.; Rose, S. A yeast two-hybrid screen reveals that osteopontin associates with MAP1A and MAP1B in addition to other proteins linked to microtubule stability, apoptosis and protein degradation in the human brain. Eur. J. Neurosci. 2012, 36, 2733–2742. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Bjerrum, J.T.; Csillag, C.; Nielsen, F.C.; Olsen, J. Influence of smoking on colonic gene expression profile in Crohn’s disease. PLoS ONE 2009, 4, e6210. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, W.; Liu, H.; Xue, M.; Liu, X.; Zhang, K.; Hu, L.; Li, J.; Liu, X.; Xiang, Z.; et al. Correction: African Swine Fever Virus pI215L Negatively Regulates cGAS-STING Signaling Pathway through Recruiting RNF138 to Inhibit K63-Linked Ubiquitination of TBK1. J. Immunol. 2022, 208, 1510–1511. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Z.; Liu, S.; Zhang, X.; Cao, X.; Jiang, M. RNF138 inhibits late inflammatory gene transcription through degradation of SMARCC1 of the SWI/SNF complex. Cell Rep. 2023, 42, 112097. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 2009, 106, 7945–7950. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. RNF138 joins the HR team. Nat. Cell Biol. 2015, 17, 1375–1377. [Google Scholar] [CrossRef]
- Lu, Y.; Han, D.; Liu, W.; Huang, R.; Ou, J.; Chen, X.; Zhang, X.; Wang, X.; Li, S.; Wang, L.; et al. RNF138 confers cisplatin resistance in gastric cancer cells via activating Chk1 signaling pathway. Cancer Biol. Ther. 2018, 19, 1128–1138. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.; Feng, M.; Yao, L.; Deng, Z.; Zao, G.; Zhou, Y.; Chen, S.; Du, Z. Downregulation of RNF138 inhibits cellular proliferation, migration, invasion and EMT in glioma cells via suppression of the Erk signaling pathway. Oncol. Rep. 2018, 40, 3285–3296. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, R.; Ying, J.; Li, X.; Jiao, T.; Guo, L.; Zhou, H.; Wang, H.; Tuersuntuoheti, A.; Liu, J.; et al. RING finger 138 deregulation distorts NF-kB signaling and facilities colitis switch to aggressive malignancy. Signal Transduct. Target. Ther. 2022, 7, 185. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Liu, C.; Fan, J.; Zou, J.; Guo, M.; Sun, G. RNF138 Downregulates Antiviral Innate Immunity by Inhibiting IRF3 Activation. Int. J. Mol. Sci. 2023, 24, 16110. https://doi.org/10.3390/ijms242216110
Zeng X, Liu C, Fan J, Zou J, Guo M, Sun G. RNF138 Downregulates Antiviral Innate Immunity by Inhibiting IRF3 Activation. International Journal of Molecular Sciences. 2023; 24(22):16110. https://doi.org/10.3390/ijms242216110
Chicago/Turabian StyleZeng, Xianhuang, Chaozhi Liu, Jinhao Fan, Jiabin Zou, Mingxiong Guo, and Guihong Sun. 2023. "RNF138 Downregulates Antiviral Innate Immunity by Inhibiting IRF3 Activation" International Journal of Molecular Sciences 24, no. 22: 16110. https://doi.org/10.3390/ijms242216110